Next: d'Auvergne protocol GUI mode Up: The new protocol in Previous: d'Auvergne protocol GUI mode Contents Index
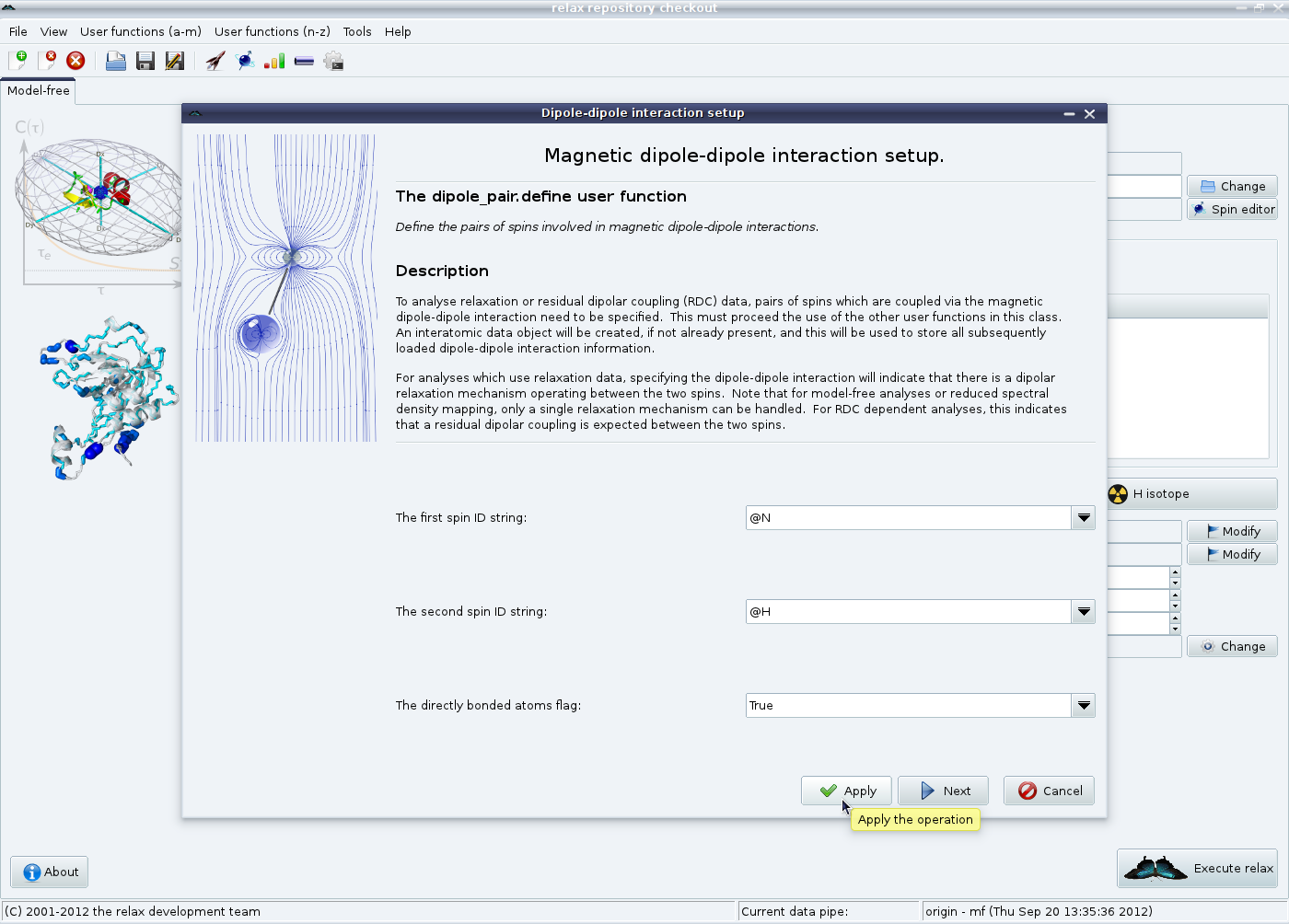
Just as in the scripting mode, the relaxation interactions need to now be defined. The first is the magnetic diole-dipole interaction. All coupled nitrogen and proton spins should already be loaded at this point. Click on the “Dipolar relaxation” button in the model-free tab in the main relax window to launch the magnetic dipole-dipole interaction wizard:
|
For this example, directly bonded nitrogens and protons will be analysed. To start with, the backbone NH pairs will be defined. Leave the values at “@N” and “@H” and click on the “Apply” button. Then change the two spin ID strings to “@NE1” and “@HE1” to set up the tryptophan sidechain indole NH pairs and click on the “Next” button. Note that the regular expression “@N*” and “@H*” should not be used in this first wizard page as otherwise @N spins will be connected to @HE1 spins of the same tryptophan residue and @H spins to @NE1 spins.
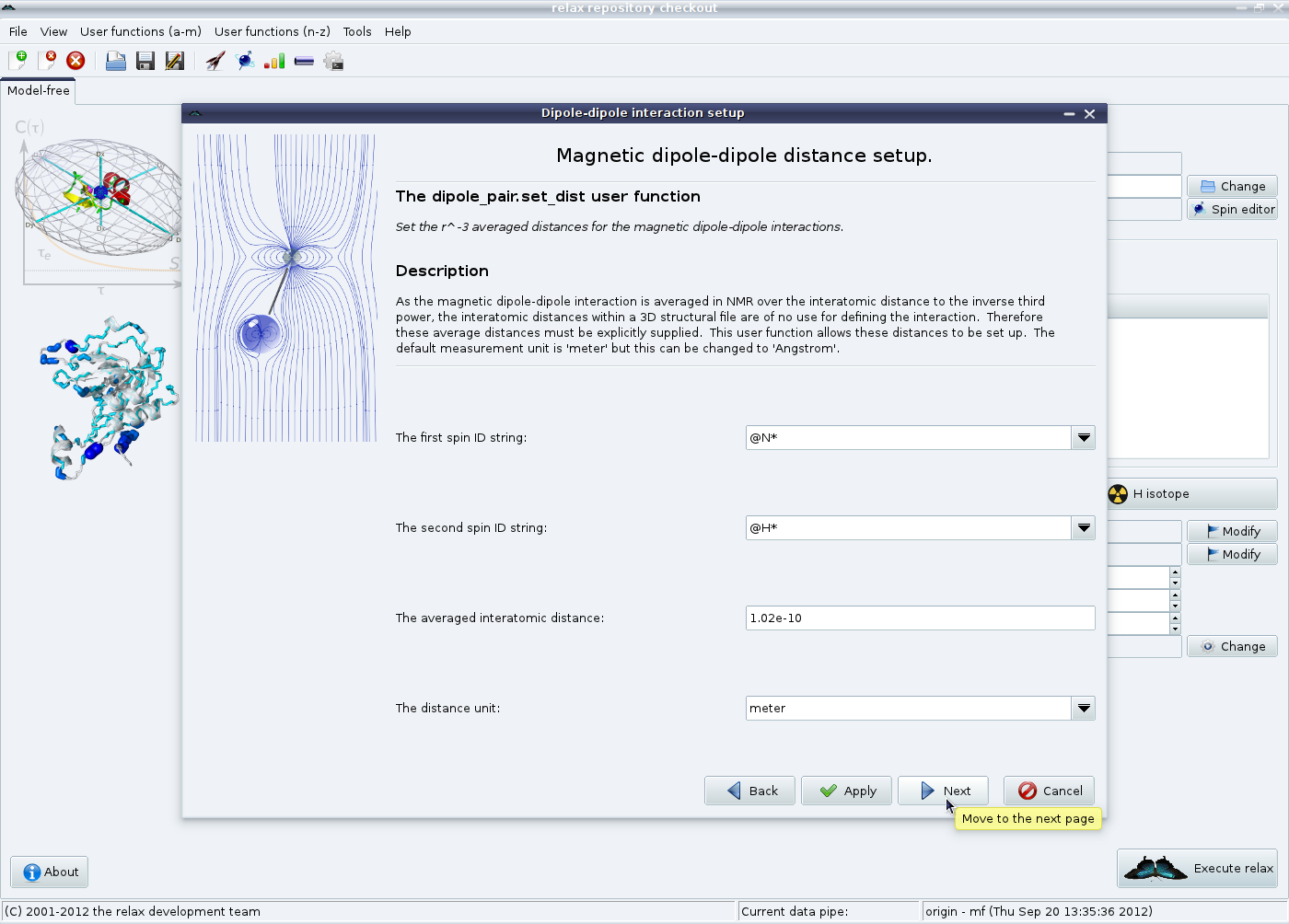
Now the 〈r-3〉 averaged distance of 1.02 Å will be set. Leave all settings as they are and click on “Next”:
|
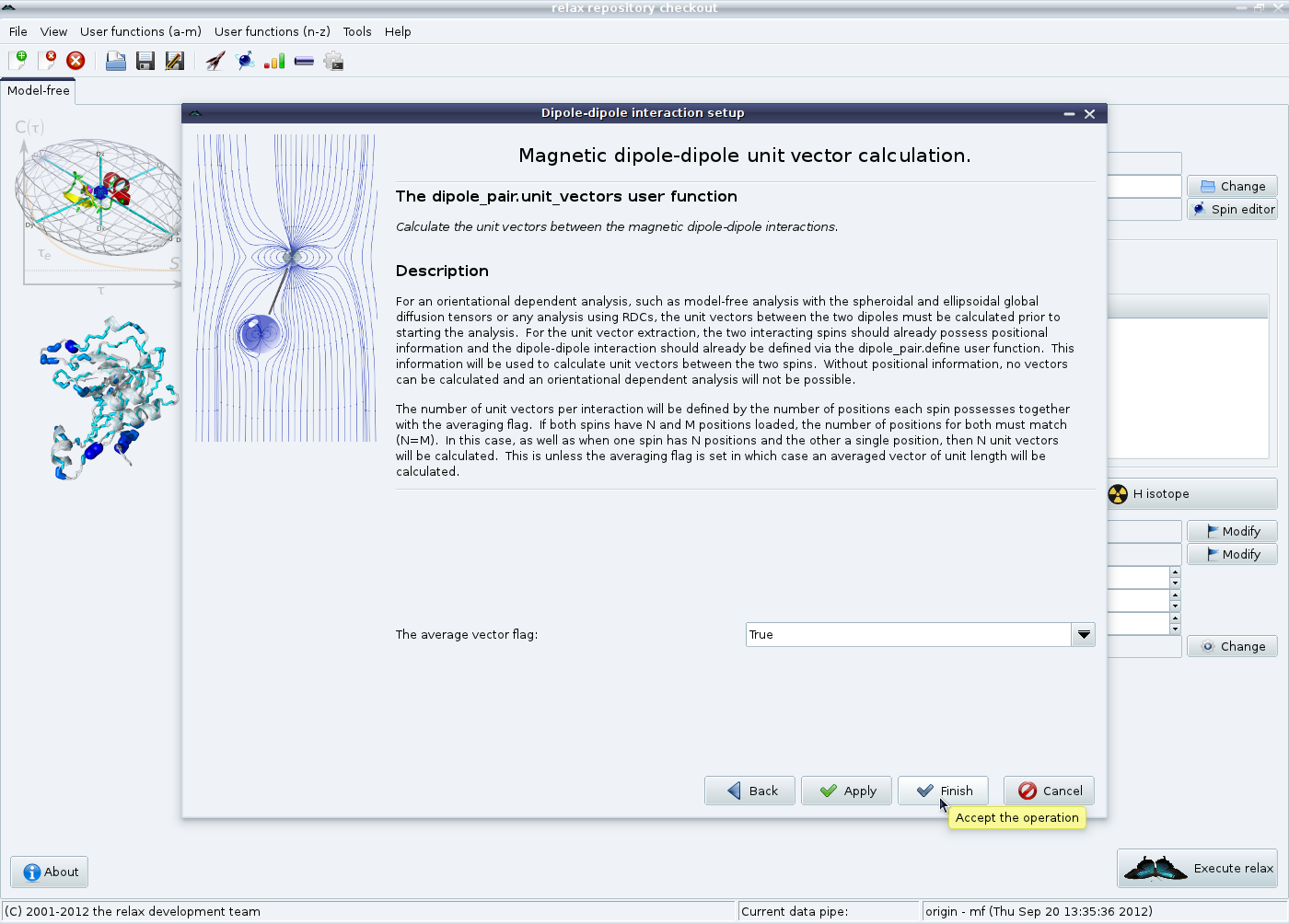
If multiple models have been loaded in the previous steps, then the unit vectors between each model need to be calculated. For a model-free analysis multiple unit vectors must be averaged to a single vector - current model-free theory is based on the assumption of a single vector orientation. Therefore the averaged vector flag must be left on “True”. Click on “Finish” to terminate the set up of the magnetic dipole-dipole interactions:
|
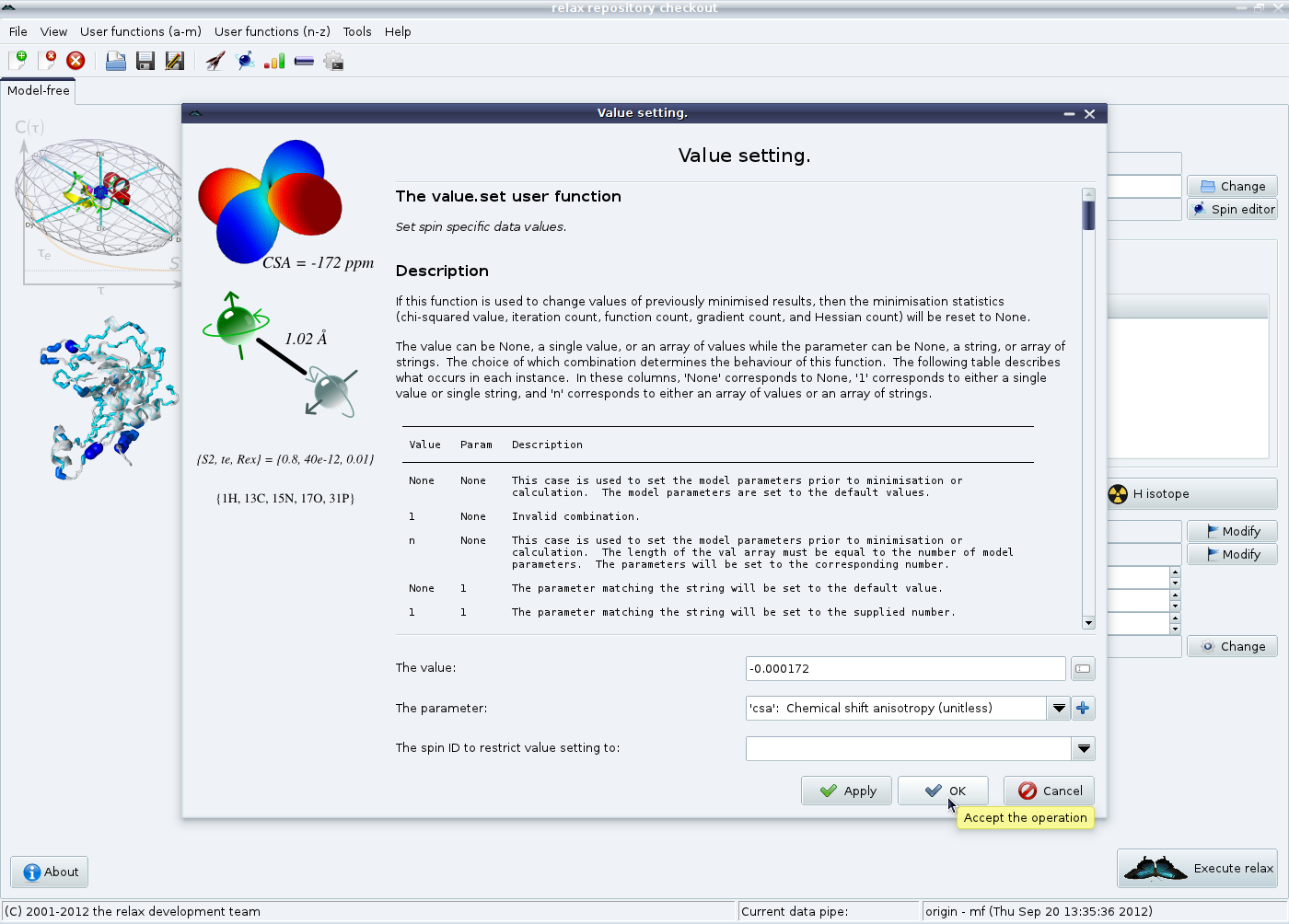
Secondly the chemical shift anisotropy (CSA) relaxation mechanism needs to be defined. Click on the “CSA relaxation” button in the model-free tab in the main relax window. An averaged CSA value of -172 ppm will be used for all spins, so simply click on “Ok” to finish.
|