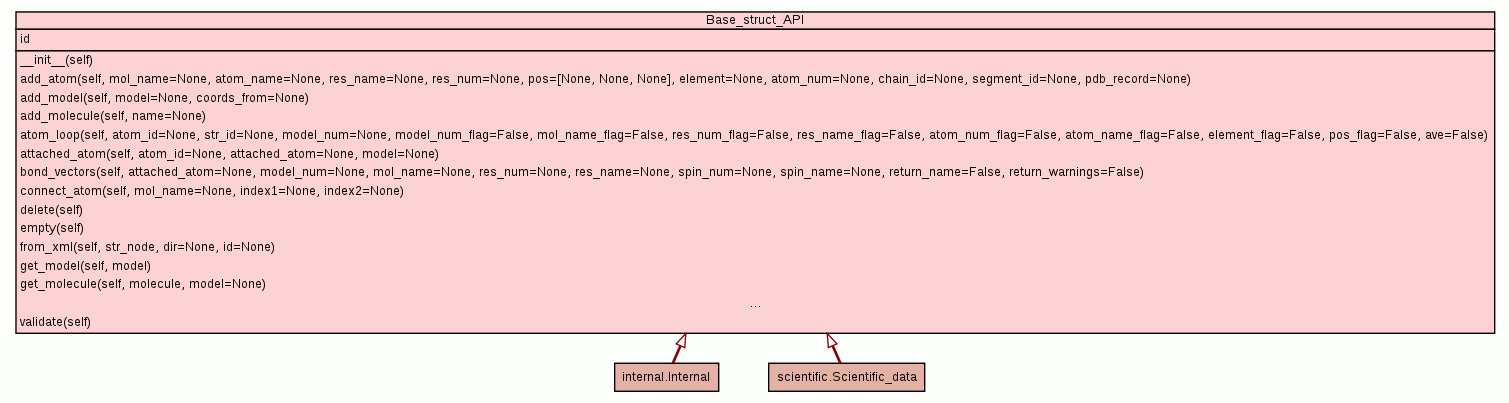
|
Prototype method stub for the creation of a PDB file from the
structural data.
The PDB records
The following information about the PDB records has been taken from
the "Protein Data Bank Contents Guide: Atomic Coordinate Entry
Format Description" version 3.1, February 11, 2008.
HET record
The HET record describes non-standard residues. The format of the
record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "HET " | |
| 8 - 10 | LString(3) | hetID | Het identifier, right-justified. |
| 13 | Character | ChainID | Chain identifier. |
| 14 - 17 | Integer | seqNum | Sequence number. |
| 18 | AChar | iCode | Insertion code. |
| 21 - 25 | Integer | numHetAtoms | Number of HETATM records for the group present |
| | | | in the entry. |
| 31 - 70 | String | text | Text describing Het group. |
|_________|______________|______________|________________________________________________|
HETNAM record
The HETNAM associates a chemical name with the hetID from the HET
record. The format is of the record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "HETNAM" | |
| 9 - 10 | Continuation | continuation | Allows concatenation of multiple records. |
| 12 - 14 | LString(3) | hetID | Het identifier, right-justified. |
| 16 - 70 | String | text | Chemical name. |
|_________|______________|______________|________________________________________________|
FORMUL record
The chemical formula for non-standard groups. The format of the
record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "FORMUL" | |
| 9 - 10 | Integer | compNum | Component number. |
| 13 - 15 | LString(3) | hetID | Het identifier. |
| 17 - 18 | Integer | continuation | Continuation number. |
| 19 | Character | asterisk | "*" for water. |
| 20 - 70 | String | text | Chemical formula. |
|_________|______________|______________|________________________________________________|
MODEL record
The model number, for multiple structures. The format of the
record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "MODEL " | |
| 11 - 14 | Integer | serial | Model serial number. |
|_________|______________|______________|________________________________________________|
ATOM record
The ATOM record contains the atomic coordinates for atoms
belonging to standard residues. The format of the record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "ATOM" | |
| 7 - 11 | Integer | serial | Atom serial number. |
| 13 - 16 | Atom | name | Atom name. |
| 17 | Character | altLoc | Alternate location indicator. |
| 18 - 20 | Residue name | resName | Residue name. |
| 22 | Character | chainID | Chain identifier. |
| 23 - 26 | Integer | resSeq | Residue sequence number. |
| 27 | AChar | iCode | Code for insertion of residues. |
| 31 - 38 | Real(8.3) | x | Orthogonal coordinates for X in Angstroms. |
| 39 - 46 | Real(8.3) | y | Orthogonal coordinates for Y in Angstroms. |
| 47 - 54 | Real(8.3) | z | Orthogonal coordinates for Z in Angstroms. |
| 55 - 60 | Real(6.2) | occupancy | Occupancy. |
| 61 - 66 | Real(6.2) | tempFactor | Temperature factor. |
| 73 - 76 | LString(4) | segID | Segment identifier, left-justified. |
| 77 - 78 | LString(2) | element | Element symbol, right-justified. |
| 79 - 80 | LString(2) | charge | Charge on the atom. |
|_________|______________|______________|________________________________________________|
HETATM record
The HETATM record contains the atomic coordinates for atoms
belonging to non-standard groups. The format of the record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "HETATM" | |
| 7 - 11 | Integer | serial | Atom serial number. |
| 13 - 16 | Atom | name | Atom name. |
| 17 | Character | altLoc | Alternate location indicator. |
| 18 - 20 | Residue name | resName | Residue name. |
| 22 | Character | chainID | Chain identifier. |
| 23 - 26 | Integer | resSeq | Residue sequence number. |
| 27 | AChar | iCode | Code for insertion of residues. |
| 31 - 38 | Real(8.3) | x | Orthogonal coordinates for X. |
| 39 - 46 | Real(8.3) | y | Orthogonal coordinates for Y. |
| 47 - 54 | Real(8.3) | z | Orthogonal coordinates for Z. |
| 55 - 60 | Real(6.2) | occupancy | Occupancy. |
| 61 - 66 | Real(6.2) | tempFactor | Temperature factor. |
| 73 - 76 | LString(4) | segID | Segment identifier; left-justified. |
| 77 - 78 | LString(2) | element | Element symbol; right-justified. |
| 79 - 80 | LString(2) | charge | Charge on the atom. |
|_________|______________|______________|________________________________________________|
TER record
The end of the ATOM and HETATM records for a chain. According to
the draft atomic coordinate entry format description:
"The TER record has the same residue name, chain
identifier, sequence number and insertion code as the terminal
residue. The serial number of the TER record is one number greater
than the serial number of the ATOM/HETATM preceding the
TER."
The format of the record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "TER " | |
| 7 - 11 | Integer | serial | Serial number. |
| 18 - 20 | Residue name | resName | Residue name. |
| 22 | Character | chainID | Chain identifier. |
| 23 - 26 | Integer | resSeq | Residue sequence number. |
| 27 | AChar | iCode | Insertion code. |
|_________|______________|______________|________________________________________________|
CONECT record
The connectivity between atoms. This is required for all HET
groups and for non-standard bonds. The format of the record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "CONECT" | |
| 7 - 11 | Integer | serial | Atom serial number |
| 12 - 16 | Integer | serial | Serial number of bonded atom |
| 17 - 21 | Integer | serial | Serial number of bonded atom |
| 22 - 26 | Integer | serial | Serial number of bonded atom |
| 27 - 31 | Integer | serial | Serial number of bonded atom |
| 32 - 36 | Integer | serial | Serial number of hydrogen bonded atom |
| 37 - 41 | Integer | serial | Serial number of hydrogen bonded atom |
| 42 - 46 | Integer | serial | Serial number of salt bridged atom |
| 47 - 51 | Integer | serial | Serial number of hydrogen bonded atom |
| 52 - 56 | Integer | serial | Serial number of hydrogen bonded atom |
| 57 - 61 | Integer | serial | Serial number of salt bridged atom |
|_________|______________|______________|________________________________________________|
ENDMDL record
The end of model record, for multiple structures. The format of
the record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "ENDMDL" | |
|_________|______________|______________|________________________________________________|
MASTER record
The control record for bookkeeping. The format of the record
is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "MASTER" | |
| 11 - 15 | Integer | numRemark | Number of REMARK records |
| 16 - 20 | Integer | "0" | |
| 21 - 25 | Integer | numHet | Number of HET records |
| 26 - 30 | Integer | numHelix | Number of HELIX records |
| 31 - 35 | Integer | numSheet | Number of SHEET records |
| 36 - 40 | Integer | numTurn | Number of TURN records |
| 41 - 45 | Integer | numSite | Number of SITE records |
| 46 - 50 | Integer | numXform | Number of coordinate transformation records |
| | | | (ORIGX+SCALE+MTRIX) |
| 51 - 55 | Integer | numCoord | Number of atomic coordinate records |
| | | | (ATOM+HETATM) |
| 56 - 60 | Integer | numTer | Number of TER records |
| 61 - 65 | Integer | numConect | Number of CONECT records |
| 66 - 70 | Integer | numSeq | Number of SEQRES records |
|_________|______________|______________|________________________________________________|
END record
The end of the PDB file. The format of the record is:
__________________________________________________________________________________________
| | | | |
| Columns | Data type | Field | Definition |
|_________|______________|______________|________________________________________________|
| | | | |
| 1 - 6 | Record name | "END " | |
|_________|______________|______________|________________________________________________|
- Parameters:
file (file object) - The PDB file object. This object must be writable.model_num (None or int) - The model to place into the PDB file. If not supplied, then all
models will be placed into the file.
|