| Trees | Indices | Help |
|
|---|
|
|
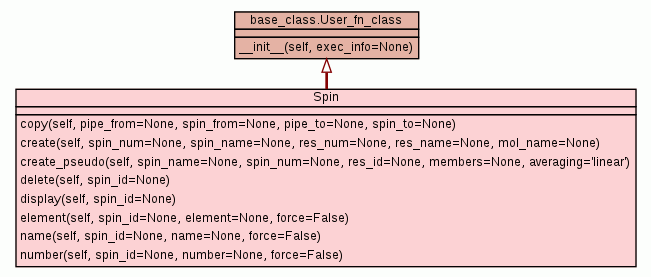
Class for manipulating the spin data.
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
Inherited from |
|||
|
|||
Copy all data associated with a spin. Keyword Arguments ~~~~~~~~~~~~~~~~~ pipe_from: The data pipe containing the spin from which the data will be copied. This defaults to the current data pipe. spin_from: The spin identifier string of the spin to copy the data from. pipe_to: The data pipe to copy the data to. This defaults to the current data pipe. spin_to: The spin identifier string of the spin to copy the data to. Description ~~~~~~~~~~~ This will copy all the data associated with the identified spin to the new, non-existent spin. The new spin must not already exist. Examples ~~~~~~~~ To copy the spin data from spin 1 to the new spin 2, type: relax> spin.copy(spin_from='@1', spin_to='@2') To copy spin 1 of the molecule 'Old mol' to spin 5 of the molecule 'New mol', type: relax> spin.copy(spin_from='#Old mol@1', spin_to='#New mol@5') To copy the spin data of spin 1 from the data pipe 'm1' to 'm2', assuming the current data pipe is 'm1', type: relax> spin.copy(spin_from='@1', pipe_to='m2') relax> spin.copy(pipe_from='m1', spin_from='@1', pipe_to='m2', spin_to='@1') |
Create a new spin. Keyword Arguments ~~~~~~~~~~~~~~~~~ spin_num: The spin number. spin_name: The name of the spin. res_num: The number of the residue to add the spin to. res_name: The name of the residue to add the spin to. mol_name: The name of the molecule to add the spin to. Description ~~~~~~~~~~~ This will add a new spin data container to the relax data storage object. The same spin number cannot be used more than once. Examples ~~~~~~~~ The following sequence of commands will generate the sequence 1 C4, 2 C9, 3 C15: relax> spin.create(1, 'C4') relax> spin.create(2, 'C9') relax> spin.create(3, 'C15') |
Create a spin system representing a pseudo-atom.
Keyword Arguments
~~~~~~~~~~~~~~~~~
spin_name: The name of the pseudo-atom spin.
spin_num: The spin number.
res_id: The molecule and residue ID string identifying the position to add the pseudo-spin to.
members: A list of the atoms (as spin ID strings) that the pseudo-atom is composed of.
averaging: The positional averaging technique.
Description
~~~~~~~~~~~
This will create a spin data container representing a number of pre-existing spin containers as a
pseudo-atom. The optional spin number must not already exist.
Examples
~~~~~~~~
The following will create the pseudo-atom named 'Q9' consisting of the protons '@H16',
'@H17', '@H18':
relax> spin.create_pseudo('Q9', members=['@H16', '@H17', '@H18'])
Spin ID string documentation
~~~~~~~~~~~~~~~~~~~~~~~~~~~~
The identification string is composed of three components: the molecule id token beginning with the
'#' character, the residue id token beginning with the ':' character, and the atom or spin system id
token beginning with the '@' character. Each token can be composed of multiple elements separated
by the ',' character and each individual element can either be a number (which must be an integer,
in string format), a name, or a range of numbers separated by the '-' character. Negative numbers
are supported. The full id string specification is '#<mol_name> :<res_id>[, <res_id>[, <res_id>,
...]] @<atom_id>[, <atom_id>[, <atom_id>, ...]]', where the token elements are '<mol_name>', the
name of the molecule, '<res_id>', the residue identifier which can be a number, name, or range of
numbers, '<atom_id>', the atom or spin system identifier which can be a number, name, or range of
numbers.
If one of the tokens is left out then all elements will be assumed to match. For example if the
string does not contain the '#' character then all molecules will match the string.
Regular expression can be used to select spins. For example the string '@H*' will select the
protons 'H', 'H2', 'H98'.
|
Delete spins. Keyword Arguments ~~~~~~~~~~~~~~~~~ spin_id: The spin identifier string. Description ~~~~~~~~~~~ This can be used to delete a single or sets of spins. See the identification string documentation below for more information. Spin ID string documentation ~~~~~~~~~~~~~~~~~~~~~~~~~~~~ The identification string is composed of three components: the molecule id token beginning with the '#' character, the residue id token beginning with the ':' character, and the atom or spin system id token beginning with the '@' character. Each token can be composed of multiple elements separated by the ',' character and each individual element can either be a number (which must be an integer, in string format), a name, or a range of numbers separated by the '-' character. Negative numbers are supported. The full id string specification is '#<mol_name> :<res_id>[, <res_id>[, <res_id>, ...]] @<atom_id>[, <atom_id>[, <atom_id>, ...]]', where the token elements are '<mol_name>', the name of the molecule, '<res_id>', the residue identifier which can be a number, name, or range of numbers, '<atom_id>', the atom or spin system identifier which can be a number, name, or range of numbers. If one of the tokens is left out then all elements will be assumed to match. For example if the string does not contain the '#' character then all molecules will match the string. Regular expression can be used to select spins. For example the string '@H*' will select the protons 'H', 'H2', 'H98'. |
Display information about the spin(s). Keyword Arguments ~~~~~~~~~~~~~~~~~ spin_id: The spin identification string. Spin ID string documentation ~~~~~~~~~~~~~~~~~~~~~~~~~~~~ The identification string is composed of three components: the molecule id token beginning with the '#' character, the residue id token beginning with the ':' character, and the atom or spin system id token beginning with the '@' character. Each token can be composed of multiple elements separated by the ',' character and each individual element can either be a number (which must be an integer, in string format), a name, or a range of numbers separated by the '-' character. Negative numbers are supported. The full id string specification is '#<mol_name> :<res_id>[, <res_id>[, <res_id>, ...]] @<atom_id>[, <atom_id>[, <atom_id>, ...]]', where the token elements are '<mol_name>', the name of the molecule, '<res_id>', the residue identifier which can be a number, name, or range of numbers, '<atom_id>', the atom or spin system identifier which can be a number, name, or range of numbers. If one of the tokens is left out then all elements will be assumed to match. For example if the string does not contain the '#' character then all molecules will match the string. Regular expression can be used to select spins. For example the string '@H*' will select the protons 'H', 'H2', 'H98'. |
Set the element type of the spin.
Keyword Arguments
~~~~~~~~~~~~~~~~~
spin_id: The spin identification string corresponding to one or more spins.
element: The IUPAC element name.
force: A flag which if True will cause the element to be changed.
Description
~~~~~~~~~~~
This allows the element type of the spins to be set.
Examples
~~~~~~~~
The set all spins of residue 1 to be carbons, type one of:
relax> spin.element('@1', 'C', force=True)
relax> spin.element(spin_id='@1', element='C', force=True)
Spin ID string documentation
~~~~~~~~~~~~~~~~~~~~~~~~~~~~
The identification string is composed of three components: the molecule id token beginning with the
'#' character, the residue id token beginning with the ':' character, and the atom or spin system id
token beginning with the '@' character. Each token can be composed of multiple elements separated
by the ',' character and each individual element can either be a number (which must be an integer,
in string format), a name, or a range of numbers separated by the '-' character. Negative numbers
are supported. The full id string specification is '#<mol_name> :<res_id>[, <res_id>[, <res_id>,
...]] @<atom_id>[, <atom_id>[, <atom_id>, ...]]', where the token elements are '<mol_name>', the
name of the molecule, '<res_id>', the residue identifier which can be a number, name, or range of
numbers, '<atom_id>', the atom or spin system identifier which can be a number, name, or range of
numbers.
If one of the tokens is left out then all elements will be assumed to match. For example if the
string does not contain the '#' character then all molecules will match the string.
Regular expression can be used to select spins. For example the string '@H*' will select the
protons 'H', 'H2', 'H98'.
|
Name the spins.
Keyword Arguments
~~~~~~~~~~~~~~~~~
spin_id: The spin identification string corresponding to one or more spins.
name: The new name.
force: A flag which if True will cause the spin to be renamed.
Description
~~~~~~~~~~~
This simply allows spins to be named (or renamed). Spin naming often essential. For example when
reading Sparky peak list files, then the spin name must match that in the file.
Examples
~~~~~~~~
The following sequence of commands will rename the sequence {1 C1, 2 C2, 3 C3} to {1 C11,
2 C12, 3 C13}:
relax> spin.name('@1', 'C11', force=True)
relax> spin.name('@2', 'C12', force=True)
relax> spin.name('@3', 'C13', force=True)
Spin ID string documentation
~~~~~~~~~~~~~~~~~~~~~~~~~~~~
The identification string is composed of three components: the molecule id token beginning with the
'#' character, the residue id token beginning with the ':' character, and the atom or spin system id
token beginning with the '@' character. Each token can be composed of multiple elements separated
by the ',' character and each individual element can either be a number (which must be an integer,
in string format), a name, or a range of numbers separated by the '-' character. Negative numbers
are supported. The full id string specification is '#<mol_name> :<res_id>[, <res_id>[, <res_id>,
...]] @<atom_id>[, <atom_id>[, <atom_id>, ...]]', where the token elements are '<mol_name>', the
name of the molecule, '<res_id>', the residue identifier which can be a number, name, or range of
numbers, '<atom_id>', the atom or spin system identifier which can be a number, name, or range of
numbers.
If one of the tokens is left out then all elements will be assumed to match. For example if the
string does not contain the '#' character then all molecules will match the string.
Regular expression can be used to select spins. For example the string '@H*' will select the
protons 'H', 'H2', 'H98'.
|
Number the spins.
Keyword Arguments
~~~~~~~~~~~~~~~~~
spin_id: The spin identification string corresponding to a single spin.
number: The new spin number.
force: A flag which if True will cause the spin to be renumbered.
Description
~~~~~~~~~~~
This simply allows spins to be numbered. The new number cannot correspond to an existing spin
number.
Examples
~~~~~~~~
The following sequence of commands will renumber the sequence {1 C1, 2 C2, 3 C3} to
{-1 C1, -2 C2, -3 C3}:
relax> spin.number('@1', -1, force=True)
relax> spin.number('@2', -2, force=True)
relax> spin.number('@3', -3, force=True)
Spin ID string documentation
~~~~~~~~~~~~~~~~~~~~~~~~~~~~
The identification string is composed of three components: the molecule id token beginning with the
'#' character, the residue id token beginning with the ':' character, and the atom or spin system id
token beginning with the '@' character. Each token can be composed of multiple elements separated
by the ',' character and each individual element can either be a number (which must be an integer,
in string format), a name, or a range of numbers separated by the '-' character. Negative numbers
are supported. The full id string specification is '#<mol_name> :<res_id>[, <res_id>[, <res_id>,
...]] @<atom_id>[, <atom_id>[, <atom_id>, ...]]', where the token elements are '<mol_name>', the
name of the molecule, '<res_id>', the residue identifier which can be a number, name, or range of
numbers, '<atom_id>', the atom or spin system identifier which can be a number, name, or range of
numbers.
If one of the tokens is left out then all elements will be assumed to match. For example if the
string does not contain the '#' character then all molecules will match the string.
Regular expression can be used to select spins. For example the string '@H*' will select the
protons 'H', 'H2', 'H98'.
|
| Trees | Indices | Help |
|
|---|
| Generated by Epydoc 3.0.1 on Wed Apr 10 13:31:53 2013 | http://epydoc.sourceforge.net |