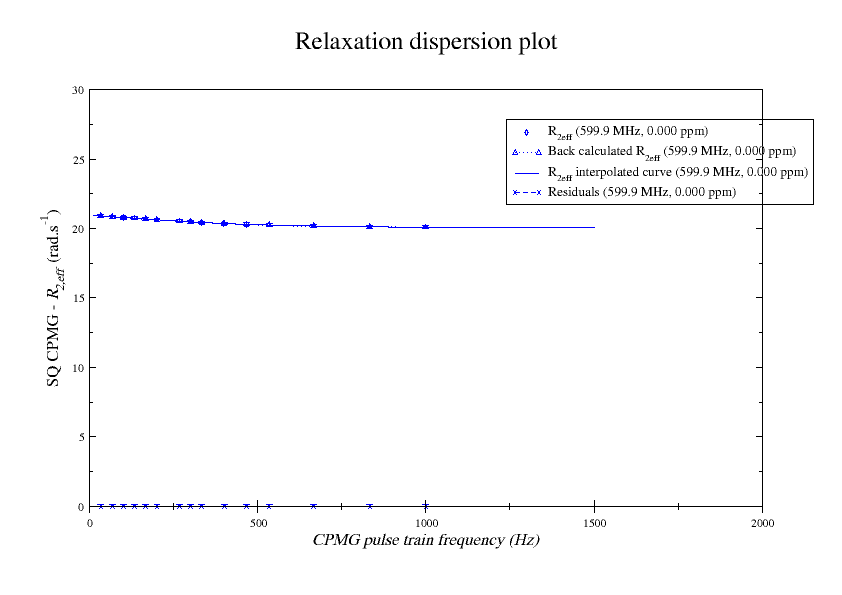
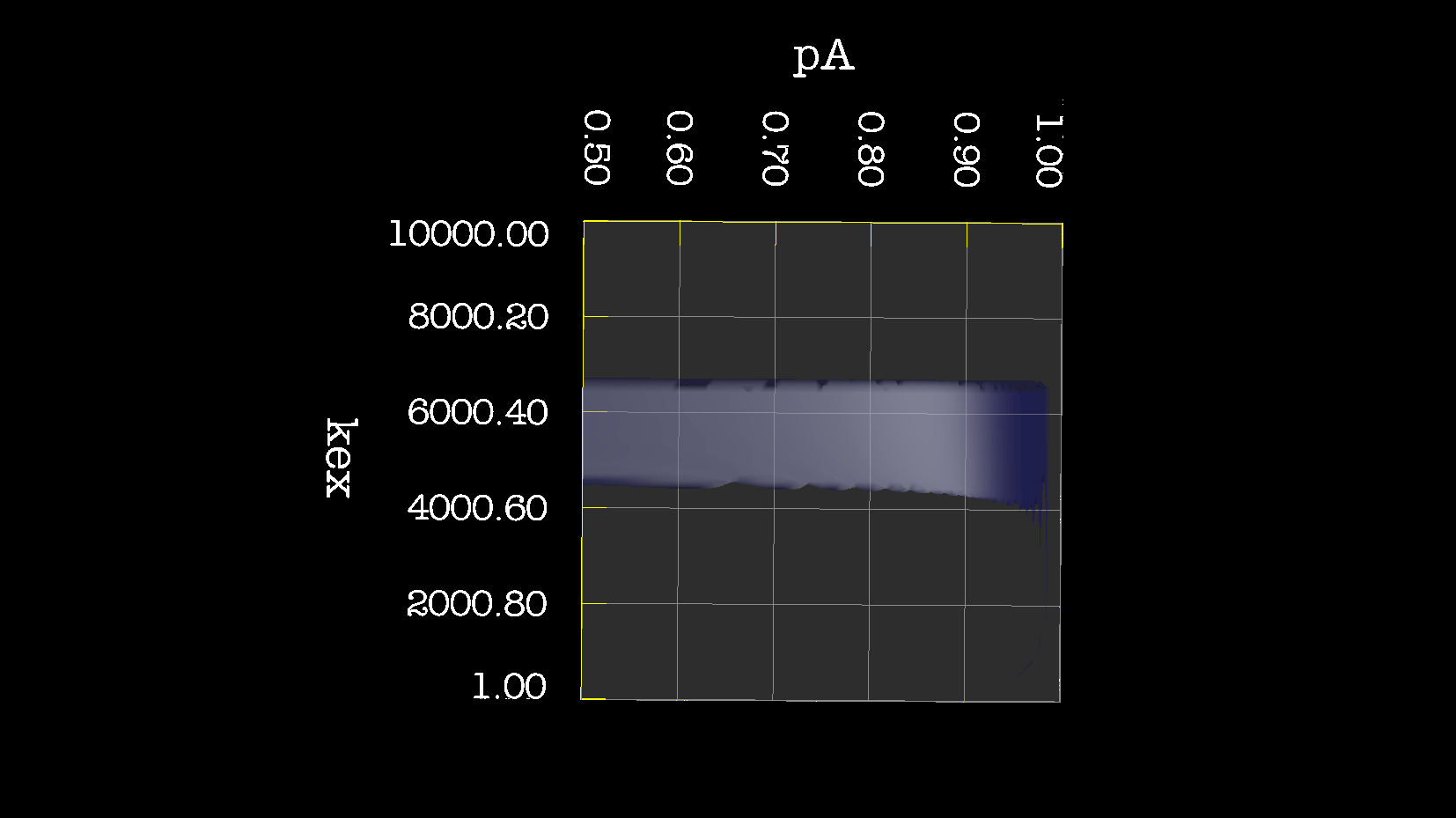
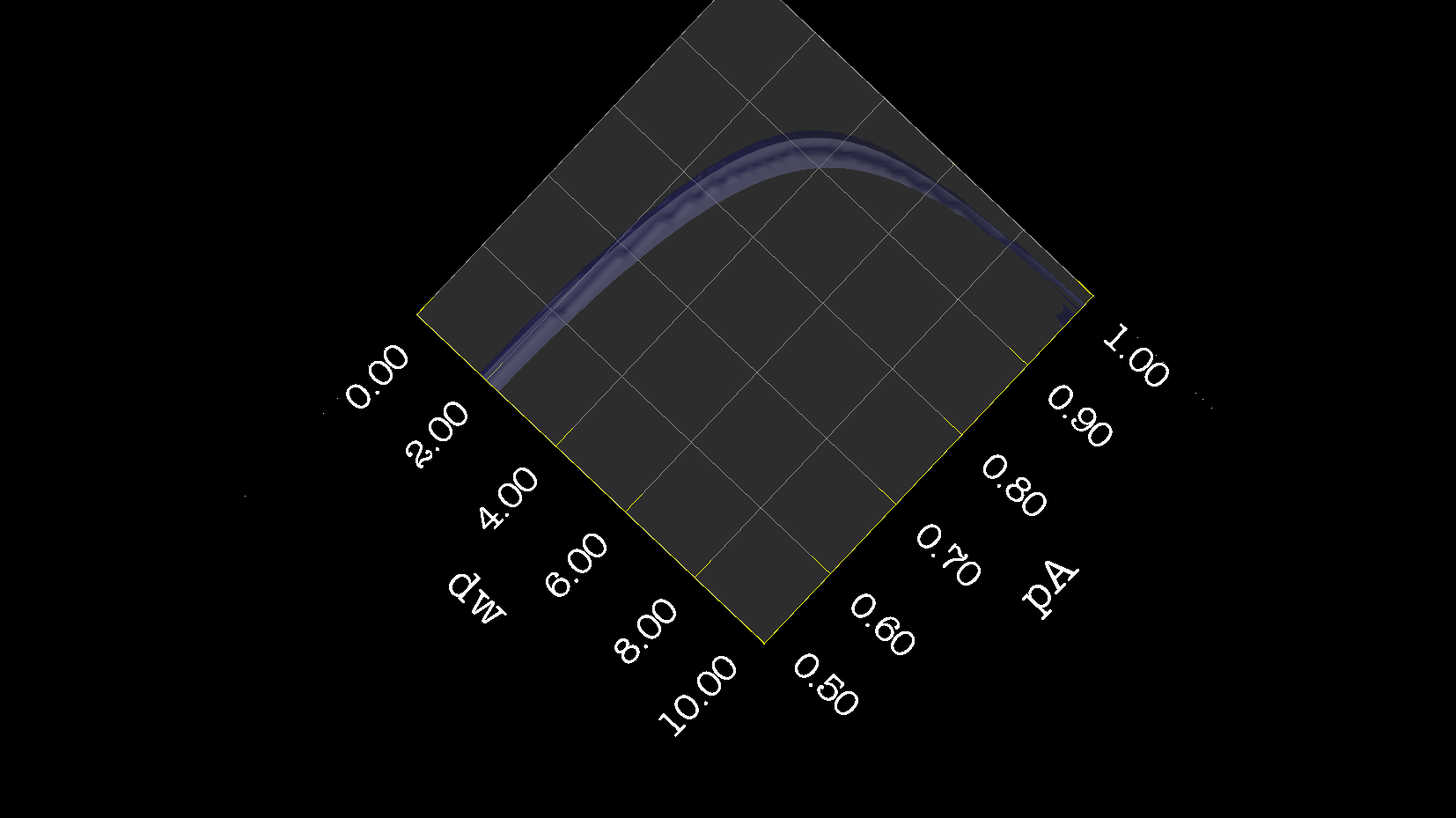
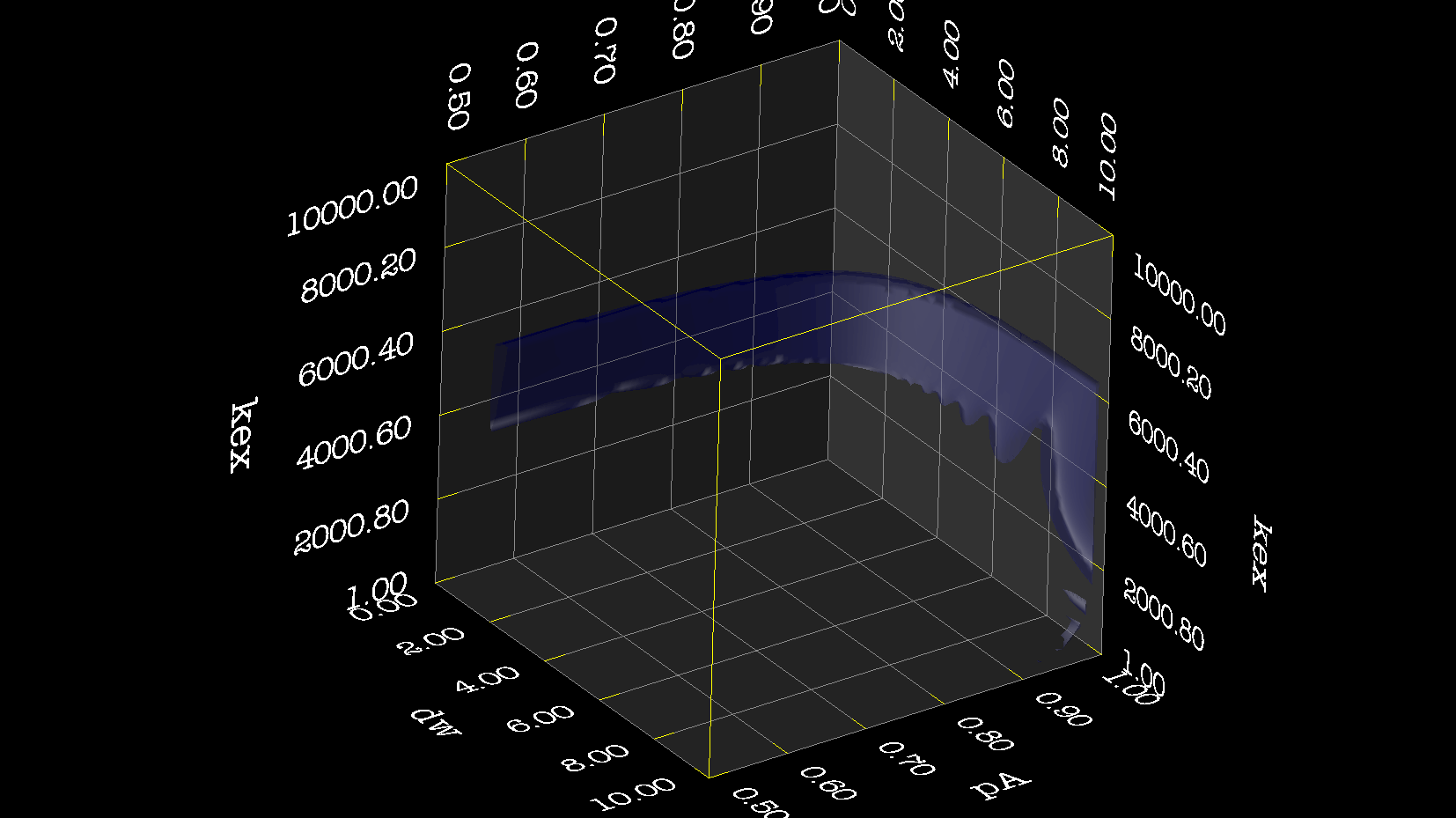
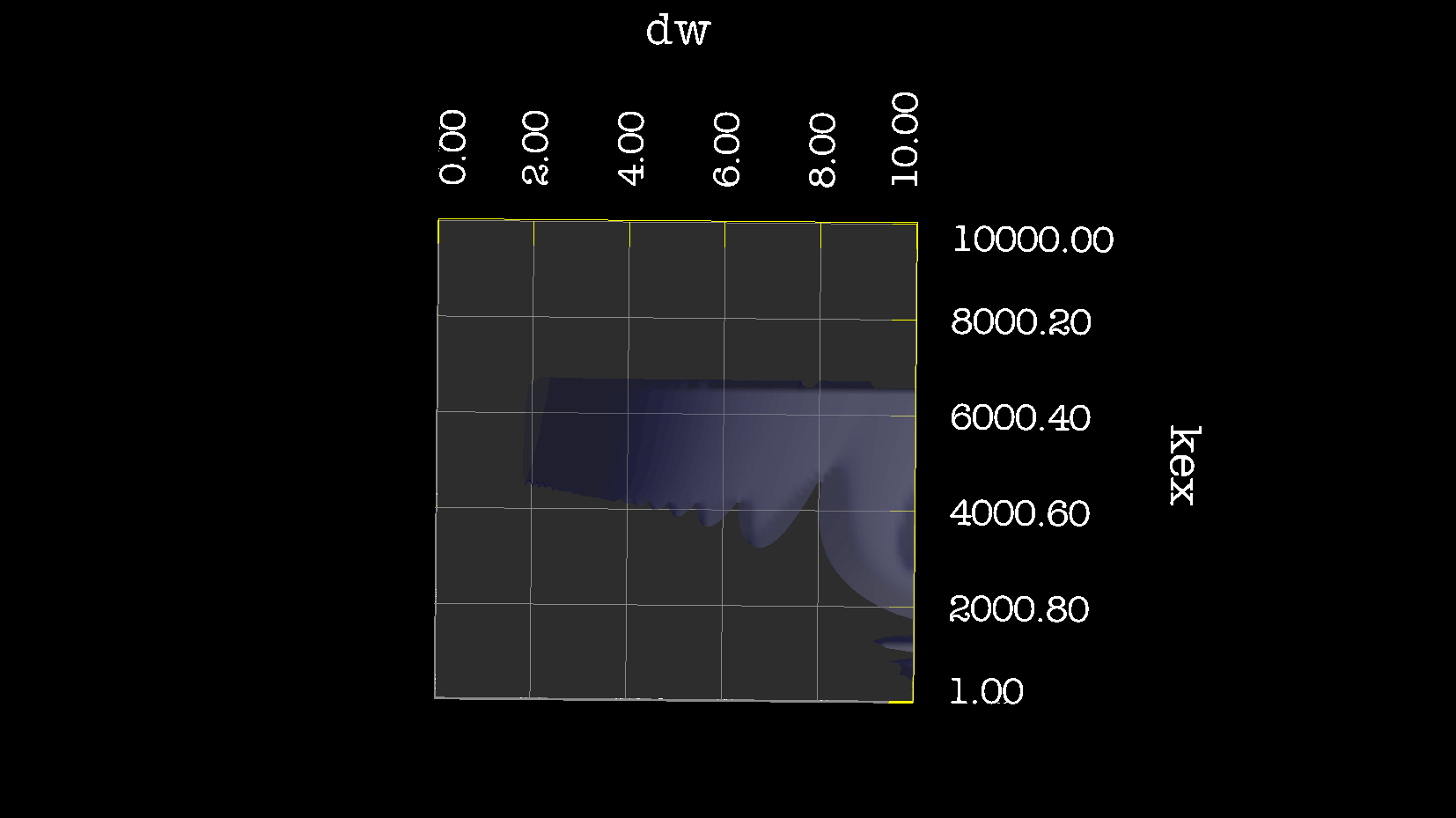
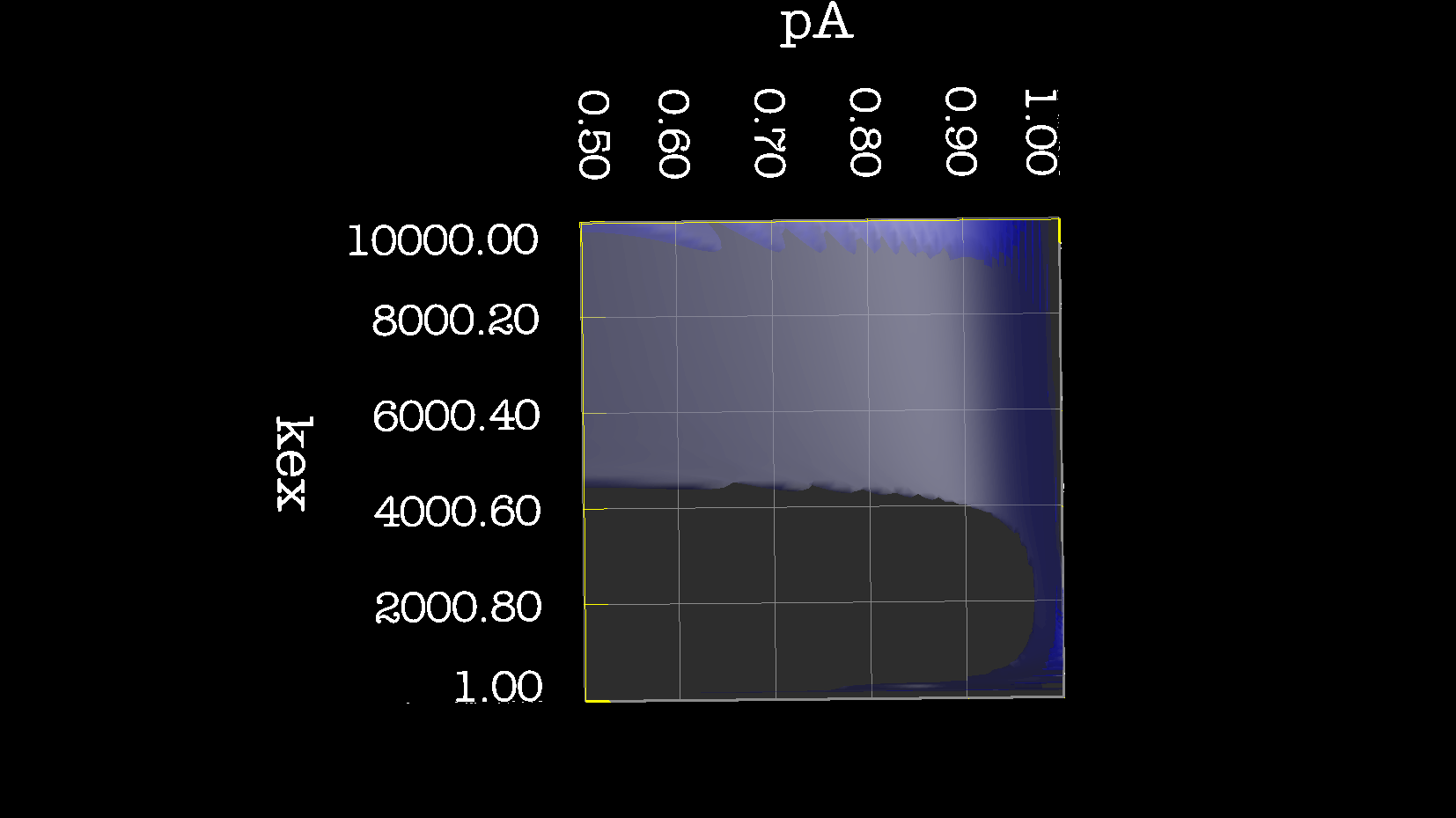
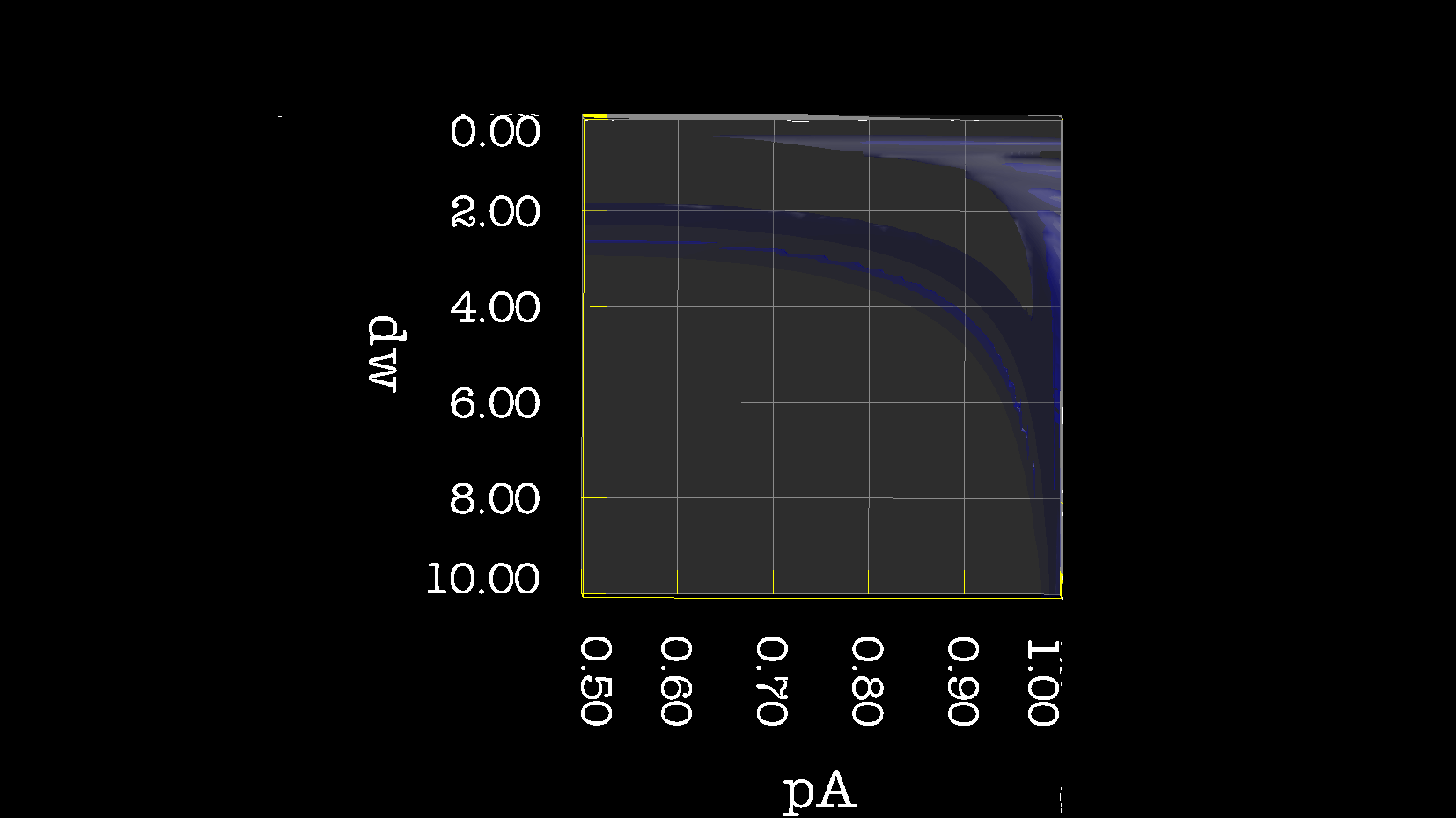
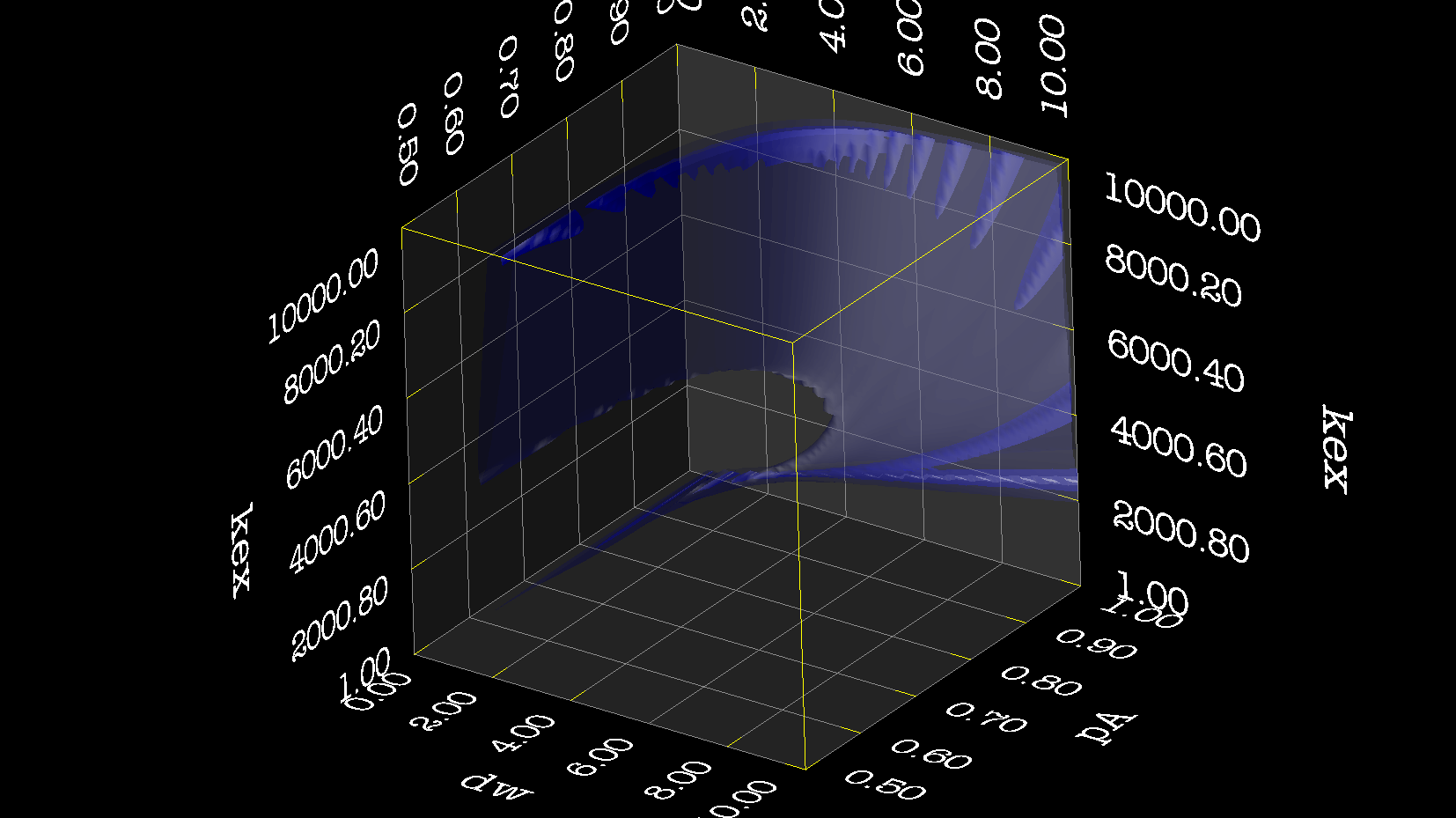
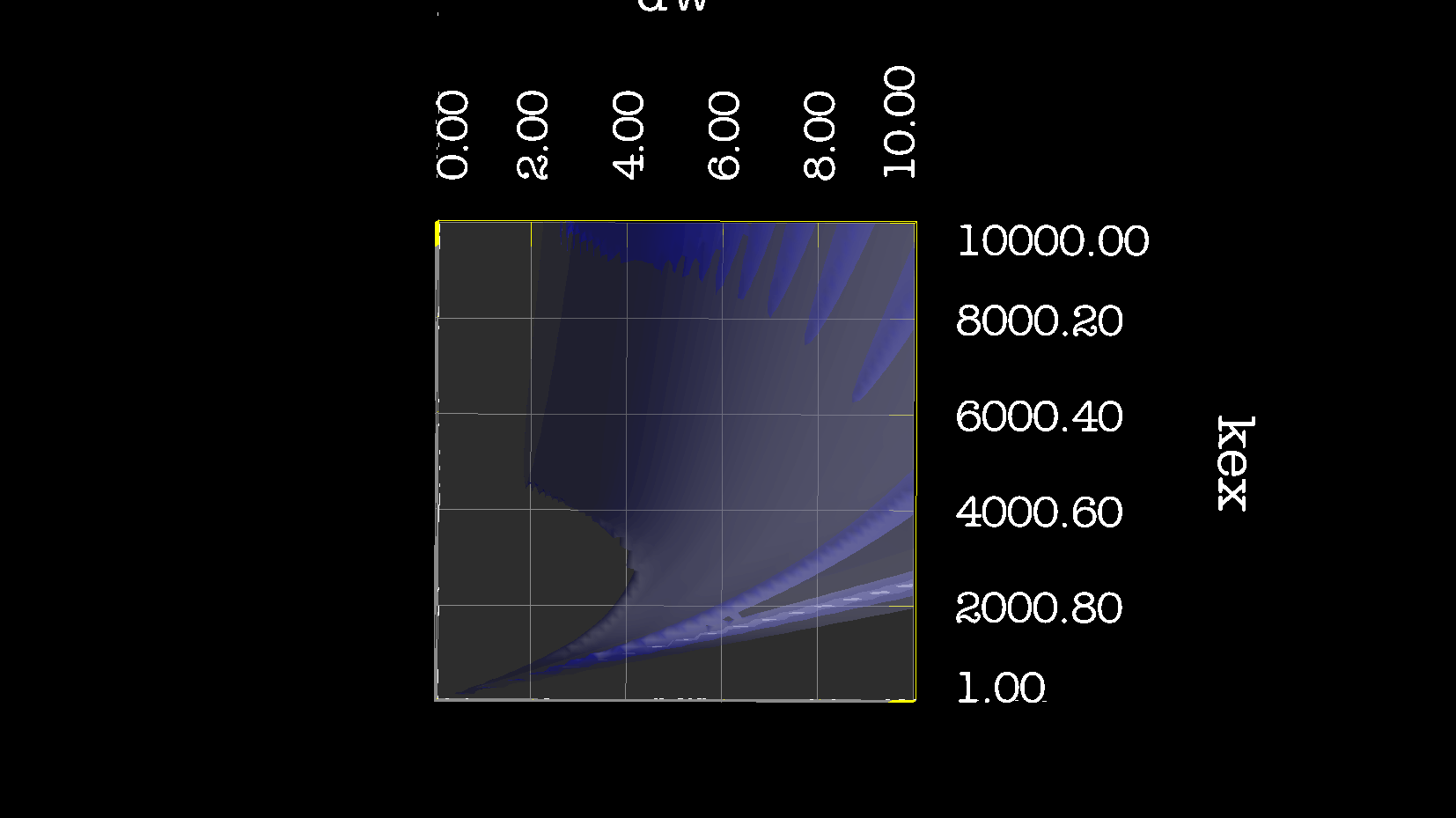
Dear Andrew. Now I know a little more about what I think. This evening, I have just discovered, why the funk, I have had extremely big problems fitting some old CPMG data with the CR72 model. It turns out, that for this system: model_create = MODEL_NS_CPMG_2SITE_EXPANDED And minimised with: model_analyse = MODEL_CR72 sfrq_1 = 599.8908617*1E6 time_T2_1 = 0.06 ncycs_1 = [28, 4, 32, 60, 2, 10, 16, 8, 20, 50, 18, 40, 6, 12, 24] sfrq_2 = 499.8908617*1E6 time_T2_2 = 0.04 ncycs_2 = [20, 16, 10, 36, 2, 12, 4, 22, 18, 40, 14, 26, 8, 32, 24, 6, 28 ] With spin dynamics created with: 'r2' = 20 rad /s. 'kex' = 2200.0 'dw' = 1.5. The Chi2 hyper surface is a !"#¤"! over the mapping of dw', 'pA', 'kex'. My problems in the fitting, has been that I have first "single minimised" 68 spins with CR72. That saves the initial 'dw', 'pA' and 'kex'. Then I have clustered these spins. The median of all pA and kex for all 68 spins, is transferred as the starting parameter. dw is kept as it is. The problem I have been inspecting, is that I saw, that sometimes relax found pA=0.5 or pA=1.0 See for example the thread: http://thread.gmane.org/gmane.science.nmr.relax.devel/5302 And then the global analysis takes FOREVER, when it is starting out with such bad parameters. Half my spins, would think they are pA=0.5, and it takes very long time to convince all 68 spins to end up at pA=0.95. It turns out, that the chi2 surface is weird for these dynamics. I have listed it is as a bug for relax! This is so weird behaviour, and have a nagging feeling something is wrong. bug #22024: (https://gna.org/bugs/index.php?22024) Minimisation space for CR72 is catastrophic. The chi2 surface over dw and pA is bounded. But chi2 surface for B14 is also very very interesting. bug #22021: (https://gna.org/bugs/index.php?22021) Model B14 shows bad fitting to data. I have used the same script to analyse the chi2 surface. Just changing the model from CR72 to B14. Take a look! I would be very grateful for any comments. Best Troels 2014-05-07 16:00 GMT+02:00 Troels Emtekær Linnet <tlinnet@xxxxxxxxxxxxx>:
Dear Andrew. I am happy to announce, that B14 is now in relax. I have 1) Optimised B14 as much as possible, Edward keeping a close eye to it! 2) Split it into two. A full with r2a!=r2b, and "normal" with r2a=r2b. 3) Documented it: # The wiki http://wiki.nmr-relax.com/B14 http://wiki.nmr-relax.com/B14_full # The library function http://svn.gna.org/viewcvs/*checkout*/relax/trunk/lib/dispersion/b14.py?revision=HEAD # The version of the manual is here at (19 MB). Page 141, table p 144, and 155. https://dl.dropboxusercontent.com/u/101911/relax_manual_with_baldwin14.pdf *Note*, that the manual now list recipe according to appendix 1, with sign conversion according to the translation from -CR72 to CR72. If you have any corrections or comments, please don't hesitate to write these.! You have also earlier expressed a wish to include some comments. We can maybe implement some lines in the manual, as: "Comments from the Author" .... That should help future NMR Spectroscopist, to make the right decision for model. If you wish, you can now also consider to change this in your manuscript. Appendix 1 – recipe for exact calculation of R2,eff "An implementation of the result written in python can be downloaded from http://baldwinlab.chem.ox.ac.uk/resources"; Just to remind you, that the code is not there yet. If you wish, you can write that it has been implemented in relax, to be released in version 3.2 http://www.nmr-relax.com/download.html With a possible reference to: http://dx.doi.org/10.1093/bioinformatics/btu166 Best Troels 2014-05-06 12:20 GMT+02:00 Troels Emtekær Linnet <tlinnet@xxxxxxxxxxxxx>:Hi Andrew. Order of my current work. 1) Optimise B14. 2) Split it into two. A full with r2a!=r2b, and "normal" with r2a=r2b. 3) Get the function code of nicolailong with r2a!=r2b into relax. 4) Modify script: http://svn.gna.org/viewcvs/*checkout*/relax/trunk/test_suite/system_tests/scripts/relax_disp/cpmg_synthetic.py?revision=HEAD 5) Make it generate data with nicolailong, fit with B14 full, CR72 full, for sensible systems. 6) Make a hyperdim space with dx: http://www.nmr-relax.com/api/3.1/pipe_control.opendx-module.html#map Then I will see what I think. Meiboom derivation is not going to happen. ;-) I am not that strong in derivations. Best Troels 2014-05-06 12:03 GMT+02:00 Andrew Baldwin <andrew.baldwin@xxxxxxxxxxxxx>:Hiya chaps, Thank you for you comments. I stand my initial statement though that the field should not use equations. Did you guys get a chance to look through the list of papers I pointed you towards? Edward, you note often that the additional physics haven't been experimentally proven to make a difference here. I think the onus is on you to read those papers, and those they cite and explain to me why they're wrong, and it's a reasonable thing to neglect these effects. You'll maybe get a 10% screw up if you use the CR equation, another 10% if you miss off resonance effects (see Ishima paper), maybe up to 3% if you miss spin flips (see Flemming's paper), and a few more percent if you're not accounting for the indirect acquisition time (see supp section 7 in the paper, and citation to Flemming). Whether or not that's a problem depends on what you're doing. A structure calculation I think is certainly broken. Getting out something fancy like an RDC or residual CSA is broken. But general trends are probably fine. People like to make the point that their kex is comparable to biochemical data from X, and make conclusions therein. I've done this myself in fact ( J. Mol. Biol. (2011) 413: 310-20). So how accurate does kex need to be before you can do that? In general, if the calculation errors are larger than your experimental errors, you're probably not doing as well as you can do. Should people revisit old data? If the data is the lynchpin of a big project, with conclusions and decisions being made from its accuracy, then probably this is a prudent thing to do...So, which of these combinations make the highest error?My fig 1 shows exactly the problems are. It's not as simple as 'this breaks in slow exchange'. One good thing about the errors in CR is that the errors scale approximately with dispersion size, so the percentage error is probably roughly constant. CR breaks most when PE is high. Note also that 'most' of your information is in the first few points of the dispersion curve, so errors here disproportionately affect the results. I think (but am not 100% sure as I don't fully understand Carver Richard's original derivation) that the PE error comes because they consider only magnetisation that originated from the ground state. Ie, if you start on the ground, hop to the excited and come back you're considered. If you start on the ground, hop to the excited, you're considered. If you start on the ground and stay there, you're considered. But if you start on the excited and jump onto the ground, you are not considered. This is very roughly what the term PD helps account for in my derivation (and it's similarly to PE is why I called it a 'P'). Setting PD to zero takes my result back to Carver Richard's.But is this Figure the same after the change in CR72 equation now?Is it really 8-10 %? for kex = 1000. and pB 5% ?. As with all things, test it yourself and see what you think :) Try it with your code if you think mine is in error. How's the Meiboom derivation coming? Best, Andy. On 06/05/2014 10:05, Troels Emtekær Linnet wrote: Dear Andrew. There is no doubt, that I think that your code is an absolutely high improvement within the field ! It is working flawless and very speedy... I am hurrying it up to implement it nicely and speedy in relax, so it can be a model to be selected in the GUI. This new release is about to come out as version 3.2 (http://www.nmr-relax.com/download.html). You new model will probably be the highlight of this release, since new model implementation is what many people really is interested in. .... I love that you make the derivation carefully, and that you take the care to describe the physical meanings. This is really something I have missed as a student, as mathematical spin operator derivations can kill me, when I can't put meaning full words on them. You paper (when typo corrected), can be a very good reference for concise derivation. Thank you for that ! So, there is no stick! There is only pure interest. :-) If your comments about the "very" bad performance of CR72 is true, I could be in trouble here. I could probably spent a year going through old dataset analysed with CR72. And really re-think if some of the biological conclusions is wrong. I can live with a "little" error, but not a catastrophic error. .... I would now like to talk a little about the results we have been reviewing. See https://gna.org/support/?3154#comment70 What we look at here, is the maximum difference in the 4D grid of kex, dw, pb, dr. ########### kexMax=1.0 #in s-1 kexMin=5000.0 dwMax=0.1 #in ppm dwMin=1000.0 pbMax=0.001 pbMin=0.1 dRMin=1. dRMax=1E4 Grid=10 For a 200 MHz. ######### So, which of these combinations make the highest error? It is known, that CR72 is "bad" in slow exchange. Slow Dynamics in Folded and Unfolded States of an SH3 Domain Tollinger, M, Skrynnikov, N R, Mulder, F a, Forman-Kay, J D, Kay, L E dx.doi.org/10.1021/ja011300z See figure 5a. (In relax as model TSMFK01: http://wiki.nmr-relax.com/TSMFK01 ) This is what your Figure 1 in your paper also should show. But is this Figure the same after the change in CR72 equation now? Is it really 8-10 %? for kex = 1000. and pB 5% ?. Best Troels 2014-05-06 10:20 GMT+02:00 Andrew Baldwin <andrew.baldwin@xxxxxxxxxxxxx>: Okay. 2 pints. And a packet of crisps :) I spent an afternoon on it and couldn't knock it into shape. It's not obvious to me what additional assumptions you need to make. Actually this true of CR result also - I couldn't work out from the paper exactly what it was that they assumed, so I set out to re-derive it. With the Meiboom, popping in the fast exachange limit is straightfoward. But then results then don't seem to boil down as nicely as you'd expect. Best, Andy. On 06/05/2014 09:07, Edward d'Auvergne wrote: Andy, you need to be a bit more fair here! If Troels can come up with a proof of the Carver and Richards 1972 equations reducing to the Luz and Meiboom 1963 equations, I would expect at least a few pints ;) Regards, Edward On 6 May 2014 09:25, Andrew Baldwin <andrew.baldwin@xxxxxxxxxxxxx> wrote: Hiya Troels, Very commendable tenacity with the fluorescence result :) Good stuff indeed! 1) Definition of alpha- : That's a fair point. I agree. Alpha- is later squared so either: zeta(B14) = 2.0*dw (Delta R2 + k_eg - k_ge) zeta(B14) = 2.0*dw (-Delta R2 - k_eg + k_ge) Will work equally well. Which you use depends on how you choose to parametrise the Eigenvalues. h1 can be plus or minus depending on your mood. So my mood says keeping the positive deltaR2 makes the equation prettier. 2) You might need to give a bit more thought to the numerical implementation. Notice that I add the factor of R2g back in, during the function Numdisp. Turns out you can factor anything out from the diagonal as long as you put it back in at the end, hence the form of the matrix in the code I sent. You can prove this by looking at the Eigenvalues, and how they propagate when you take things like matrix exponentials. In the same vein, notice that In the paper I subtract R2G from both f00 and f11. I add it back in again later in the constants out the front. This is following from the lead of Nikolai in his reconstructing invisible states paper. Implicitly looking at deltaR2s shows clearly where differences in relaxation have effects, so this brings out the physics. So look at the 'error' reported in the outputs. When you made the Troels version of the numerical code, the error for the Baldwin formula was 10s-1. This is actually the exact value you set R2G to. So while you added in R2G in the evolution matrix (which is not wrong to do), you didn't then remove it in numdisp, so your solution is out by one unit of R2G. Looking at the outputs, the code I sent for CR had the carver richard's formula set in the wrong 'minus' configuration, added during testing while writing the previous email. This was certainly my foobar. However, note that the maximum 'error' reported by the code was 4989 s-1 when you ran the code for the first time. In my previous email: with the alpha- sign made consistent with the Carver Richard's paper gives an error of 4989 s-1 in the same test That should have set off alarm bells for you. Setting either: zeta=2*dw*(deltaR2+keg-kge) #zeta=g1 zeta=2*dw*(-deltaR2-keg+kge) #zeta=g1 In the CR function as you note (and as you do in relax) is good. If you subtract the 'Baldwin error' from the 'CR error' you get 8. 8 s-1 is the biggest error incurred using CR in this parameter range (as I stated in the last email). So it's fair to call the CR equation an approximation. Fig 1 speaketh the truth. Calculate it yourself first using your code before wielding the accusing stick! So I'm deducting one half pint from your score for not checking the results from your modified numerical code more carefully. And now for beer counting: 1) Appendix 1 – recipe for exact calculation of R2,eff is: h1 = 2.0*dw (R2B - R2A + k_BA - k_AB) = - CR72 (But it have no influence, but please keep history! Change it back to CR72. :-)) same for h2. Pint for the typo in keg/kge. That's a genuine foobar in the manu - the equations should match code i've written, and I know the code is right because i've matched it to simulation. I'll keep my Eigenvalue definitions though. 2) zeta typo in eq 70. z=zeta Agreed! +1. 3) Watch alpha_ in eq 70. Get Delta R straight See 1. No extra pint here. 4) Comments in CODE: http://svn.gna.org/viewcvs/*checkout*/relax/trunk/lib/dispersion/b14.py?revision=HEAD g1=2*dw*(deltaR2+keg-kge) #same as carver richards zeta That is not true. That is minus Zeta. But it does not matter, since order **2. See 1. No extra pint here either. 5) Did you actually implement ExchangeMat2x2 wrong?? If yes, theeeeeen, no wonder figure 1 shows what it does. :-) What is up for the deltaOmegas story??? -1/2 pint for an over-reached conclusion from your modified code. So current score = 1.5 Meiboom challenge: I'd be interested in a proof for how Carver Richard's reduces to Meiboom. I'd pay a pint for that. To get you started, note that in the fast exchange limit, E0 and E2 can be simplified using the fast exchange limits in supp section 1. Best, Andy. On 05/05/2014 15:27, Troels Emtekær Linnet wrote: Hi Andy. Heh, the funniest part with the fluorescence was to battle with the moderators of wikipedia to change 9000 to 9 in R06. http://en.wikipedia.org/wiki/F%C3%B6rster_resonance_energy_transfer#Theoretical_basis That was a brawl, to convince them that the bible was wrong. But referring to the old dusty library book of Forster (Ref 14) made the deal. Back to the beer. You are swapping two things in your code, according to CR72. If I write up CR72: (Directly from cr72.py) ####### fact = r20a - r20b - k_BA + k_AB zeta = 2.0*dw * fact So in total: %%%%% zeta(CR72) = 2.0*dw (R2A - R2B + k_AB - k_BA) %%%% Then we take your code: ####### deltaR2=R2e-R2g g1=2*dw*(deltaR2+keg-kge) #same as carver richards zeta Change notation, to make it easier to see: %%%%% zeta(B14) = 2.0*dw (R2B - R2A + k_BA - k_AB) %%%% But huh, is that not just: zeta(B14) = - zeta(CR72) = 2.0*dw ( - R2A + R2B - k_AB + k_BA) = 2.0*dw ( R2B - R2A + k_BA - k_AB) Well, that is interesting. So your g1 is the negative of carver richards zeta. Fortunately we are lucky, that zeta is to the power **2. So that leaves it out. How about psi? It boils down to that fact(B14) = - fact(CR72) <=> alpha_ (B14) = - alpha_ (CR72) But alpha_ is also to the power **2. Phew... So what is up. If we look at: (compareTroells.py) https://gna.org/support/download.php?file_id=20636 ############## def CarverRichards(kex,pb,dw,ncyc,Trelax,R2g,R2e,outfile,verb='n'): deltaR2=R2e-R2g keg=(1-pb)*kex kge=pb*kex zeta=2*dw*(-deltaR2+keg-kge) #zeta=g1 psi=(deltaR2+keg-kge)**2.0+4*keg*kge-dw**2 #psi=g2 ############# Well here we have something ! zeta = 2.0 * dw ( R2A - R2B - k_AB + k_BA ) So this is not about how you make the difference of ( R2A - R2B ) it is the swapping of + k_AB - k_BA << - k_AB + k_BA kex(1 - 2pa) << kex( -1 + 2pa ) You have implemented CR72 wrong ? Hmmmm. What is up? You get a difference between CR72, and numerical. You cannot get CR72 to work, unless you modify alpha_. You invert the signs of forward / backward exchange rate. You say that numerical is true, so CR72 is wrong. Lets inspect numerical. If we look at: (compareTroells.py) https://gna.org/support/download.php?file_id=20636 Now we look up the numerical solution. ##### NumDisp(kex,pb,dw,ncyc,Trelax,R2g,R2e,outfile,verb='n'): array=CPMG2x2(dw,(R2e-R2g),kex,pb,Trelax,ncyc) CPMG2x2(dOmega,DeltaR2,kex,pb,Trel,ncyc): L=ExchangeMat2x2(dOmega,DeltaR2,kex,pb) ExchangeMat2x2(dOmega,DeltaR2,kex,pb): L = mat(zeros((2, 2),dtype=complex)) L[1,1] -= DeltaR2 Hmmm. According to your paper at equation 3. R+ = [[ -kAB - R2A] [kBA] ] [ kAB] [ -kBA - R2B - i dw] ] But I see that it gets populated like R+ = [[ -kAB ] [kBA] ] [ kAB] [ -kBA - R2B + R2A - i dw] ] Has this something to do with it: ???? So I took the liberty to change your code, hoping the Theory is right. ############# Normal Baldwin 1.183188 1.0 Numerical 58.556647 49.4905687008 Meiboom 0.391873 0.331200958766 Carver 0.693676 0.586277075156 compareTroells_w_Troels_mod.py:260: RuntimeWarning: invalid value encountered in log R2eff=(1/Tc)*numpy.log(intensity0/intensity); # we did not factor out (Ka+Kb)/2 here Nikolai 3.434938 2.90312105938 NikolaiLong 585.485493 494.837247335 Maximum error over grid (s-1): Meiboom: 93015865.9296 Baldwin: 1.49539403083e-09 CarverRichards: 4989.8296812 Nikolai dougle (9): 204.761587913 Nikolai long double (23): 2.0692254415912185547869e-7 ############ Modifying Meiboom to 4 NCYC Maximum error over grid (s-1): Meiboom: 22664952.3388 Baldwin: 1.49539403083e-09 CarverRichards: 4989.8296812 ########### Modifying ExchangeMat2x2 to your paper version. Meiboom: 22664942.3388 Baldwin: 10.0000000012 CarverRichards: 4979.8296812 ########## Modify CR72 zeta=2*dw*(-deltaR2-keg+kge) Baldwin 1.216869 1.0 Numerical 60.302727 49.555644034 Meiboom 0.397395 0.326571718073 Carver 0.700189 0.575402118059 compareTroells_w_Troels_mod.py:261: RuntimeWarning: invalid value encountered in log R2eff=(1/Tc)*numpy.log(intensity0/intensity); # we did not factor out (Ka+Kb)/2 here Nikolai 3.569577 2.93341107383 NikolaiLong 591.459824 486.050531323 Maximum error over grid (s-1): Meiboom: 22664942.3388 Baldwin: 10.0000000012 CarverRichards: 18.2238852463 Nikolai dougle (9): 214.761587913 Nikolai long double (23): 10.000000207062814176945 WOOOOOWWWW The change I made was: ############################################## 37c37 < def ExchangeMat2x2(dOmega,DeltaR2,kex,pb): --- def ExchangeMat2x2(dOmega,R2e,R2g,kex,pb): 47c47 < L[1,1] -= DeltaR2 --- L[1,1] -= R2e 49a50 L[0, 0] -= R2g 64,65c65,66 < def CPMG2x2(dOmega,DeltaR2,kex,pb,Trel,ncyc): < L=ExchangeMat2x2(dOmega,DeltaR2,kex,pb) --- def CPMG2x2(dOmega,R2e,R2g,kex,pb,Trel,ncyc): L=ExchangeMat2x2(dOmega,R2e,R2g,kex,pb) 86,87c87,88 < tcp=1/(2*nu_cpmg) < R2eff=(1-pb)*R2g+pb*R2e+R2Fast*(1.-(2./(kex*tcp))*numpy.tanh(kex*tcp/2.0)) --- tcp=1/(4*nu_cpmg) R2eff=(1-pb)*R2g+pb*R2e+R2Fast*(1.-(4./(kex*tcp))*numpy.tanh(kex*tcp/4.0)) 112c113 < zeta=2*dw*(-deltaR2+keg-kge) #zeta=g1 --- zeta=2*dw*(-deltaR2-keg+kge) #zeta=g1 145c146 < array=CPMG2x2(dw,(R2e-R2g),kex,pb,Trelax,ncyc) --- array=CPMG2x2(dw,R2e,R2g,kex,pb,Trelax,ncyc) 546c547 < #tom,jarr=TestDisp(ncyc,Trelax,R2g,'NikolaiLong',tim) --- tom,jarr=TestDisp(ncyc,Trelax,R2g,'NikolaiLong',tim) 560c561 < print 'Nikolai long double (18): ',numpy.max(numpy.abs(narr[:,:,1]-larr[:,:,1])) --- #print 'Nikolai long double (18): ',numpy.max(numpy.abs(narr[:,:,1]-larr[:,:,1])) [tlinnet@tomat ~/Downloads]$ diff compareTroells.py compareTroells_w_Troels_mod.py 37c37 < def ExchangeMat2x2(dOmega,DeltaR2,kex,pb): --- def ExchangeMat2x2(dOmega,R2e,R2g,kex,pb): 47c47 < L[1,1] -= DeltaR2 --- L[1,1] -= R2e 49a50 L[0, 0] -= R2g 64,65c65,66 < def CPMG2x2(dOmega,DeltaR2,kex,pb,Trel,ncyc): < L=ExchangeMat2x2(dOmega,DeltaR2,kex,pb) --- def CPMG2x2(dOmega,R2e,R2g,kex,pb,Trel,ncyc): L=ExchangeMat2x2(dOmega,R2e,R2g,kex,pb) 86,87c87,88 < tcp=1/(2*nu_cpmg) < R2eff=(1-pb)*R2g+pb*R2e+R2Fast*(1.-(2./(kex*tcp))*numpy.tanh(kex*tcp/2.0)) --- tcp=1/(4*nu_cpmg) R2eff=(1-pb)*R2g+pb*R2e+R2Fast*(1.-(4./(kex*tcp))*numpy.tanh(kex*tcp/4.0)) 112c113 < zeta=2*dw*(-deltaR2+keg-kge) #zeta=g1 --- zeta=2*dw*(-deltaR2-keg+kge) #zeta=g1 145c146 < array=CPMG2x2(dw,(R2e-R2g),kex,pb,Trelax,ncyc) --- array=CPMG2x2(dw,R2e,R2g,kex,pb,Trelax,ncyc) 546c547 < #tom,jarr=TestDisp(ncyc,Trelax,R2g,'NikolaiLong',tim) --- tom,jarr=TestDisp(ncyc,Trelax,R2g,'NikolaiLong',tim) 560c561 < print 'Nikolai long double (18): ',numpy.max(numpy.abs(narr[:,:,1]-larr[:,:,1])) --- #print 'Nikolai long double (18): ',numpy.max(numpy.abs(narr[:,:,1]-larr[:,:,1])) [tlinnet@tomat ~/Downloads]$ diff compareTroells.py compareTroells_w_Troels_mod.py 37c37 < def ExchangeMat2x2(dOmega,DeltaR2,kex,pb): --- def ExchangeMat2x2(dOmega,R2e,R2g,kex,pb): 47c47 < L[1,1] -= DeltaR2 --- L[1,1] -= R2e 49a50 L[0, 0] -= R2g 64,65c65,66 < def CPMG2x2(dOmega,DeltaR2,kex,pb,Trel,ncyc): < L=ExchangeMat2x2(dOmega,DeltaR2,kex,pb) --- def CPMG2x2(dOmega,R2e,R2g,kex,pb,Trel,ncyc): L=ExchangeMat2x2(dOmega,R2e,R2g,kex,pb) 86,87c87,88 < tcp=1/(2*nu_cpmg) < R2eff=(1-pb)*R2g+pb*R2e+R2Fast*(1.-(2./(kex*tcp))*numpy.tanh(kex*tcp/2.0)) --- tcp=1/(4*nu_cpmg) R2eff=(1-pb)*R2g+pb*R2e+R2Fast*(1.-(4./(kex*tcp))*numpy.tanh(kex*tcp/4.0)) 112c113 < zeta=2*dw*(-deltaR2+keg-kge) #zeta=g1 --- zeta=2*dw*(-deltaR2-keg+kge) #zeta=g1 145c146 < array=CPMG2x2(dw,(R2e-R2g),kex,pb,Trelax,ncyc) --- array=CPMG2x2(dw,R2e,R2g,kex,pb,Trelax,ncyc) 546c547 < #tom,jarr=TestDisp(ncyc,Trelax,R2g,'NikolaiLong',tim) --- tom,jarr=TestDisp(ncyc,Trelax,R2g,'NikolaiLong',tim) 560c561 < print 'Nikolai long double (18): ',numpy.max(numpy.abs(narr[:,:,1]-larr[:,:,1])) --- #print 'Nikolai long double (18): ',numpy.max(numpy.abs(narr[:,:,1]-larr[:,:,1])) #################################### File can be downloaded from: https://gna.org/support/download.php?file_id=20642 What have we (maybe) learned????? 1) Baldwin has created a code, which is as good as the numerical of Nicolai. Thanks !!! Wuhuuu ! 2) CR72 is still valid though. Doing well. And now for beer counting: 1) Appendix 1 – recipe for exact calculation of R2,eff is: h1 = 2.0*dw (R2B - R2A + k_BA - k_AB) = - CR72 (But it have no influence, but please keep history! Change it back to CR72. :-)) same for h2. 2) zeta typo in eq 70. z=zeta 3) Watch alpha_ in eq 70. Get Delta R straight 4) Comments in CODE: http://svn.gna.org/viewcvs/*checkout*/relax/trunk/lib/dispersion/b14.py?revision=HEAD g1=2*dw*(deltaR2+keg-kge) #same as carver richards zeta That is not true. That is minus Zeta. But it does not matter, since order **2. g2=(deltaR2+keg-kge)**2.0+4*keg*kge-dw**2 #same as carver richards psi Not totally correct, but since deltaR2 is swapped, it gets minus alpha, and goes to order 2. 5) Did you actually implement ExchangeMat2x2 wrong?? If yes, theeeeeen, no wonder figure 1 shows what it does. :-) What is up for the deltaOmegas story??? This is my 2 cents. :-) Let me hear if my hours have been wasted. Best Troels 2014-05-05 11:00 GMT+02:00 Andrew Baldwin <andrew.baldwin@xxxxxxxxxxxxx>: I had to struggle long to find out the Forster distance calculation in Lakowicz was wrong. The bible of fluorescence... Very good stuff :) Most would not take the time to check :) The finest biophysics really does demand both first rate biology and first rate physics. I don't think anyone has ever used the CR equation to analyse data outside of the limit where R2A=R2B, so it's very unlikely that this has error has had any real consequence. Note also that in general, it's actually much easier to code up a numerical solution - ie once you've typed up an evolution matrix, all you have left to do is write some lines taking matrix exponentials. Whereas with an equation, not only are you making implicit approximations that aren't necessarily obvious, there are many, many, more lines to type, thus increasing the probability of a foobar. So numerical solutions for spin dynamics = no mistakes, and also easy to code! This has been the approach in the Kay group, arguably cultivated by Dimitri and Nikolai, and then pushed hard more recently by Flemming, Pramodh and Guillaume. This paper was truly groundbreaking (in my view): Journal of biomolecular NMR. 2000 18(1):49-63 On this note, there are a few suggestions that Flemming and I are putting together for another thread sensibly started by Edward a few days ago for analysing R1rho and CPMG data. I think that with just a few extra tweaks, the backend to your code can be as powerful and as versatile as your front end, for millisecond data analysis. I agree that it really is in the interests of the field to have people able to perform the experiments and get information out of their data as efficiently (and as rigorously) as possible. This means more citations for those that made the experiments, and the community can keep showing more examples where Xray people have inadvertently missed functionally relevant conformers :) Best, Andy. On 04/05/2014 23:09, Troels Emtekær Linnet wrote: Well Andrew. Your paper popped out of the Mail alerts. :-) My supervisor Kaare Teilum guided my attention to it, and asked me to go through it to check. I discussed it with a lab colleague, and we were thrived. The math derivations still kills me, but I thought that the essential results was alarming. I mean, half of our lab do CPMG or R1rho. Any changes to those equations gets our attention. The "worst" problem we have in our lab, is the ability to analyse data. With relax, we now have a GUI, which make sure that at least the Bachelor Students still survive. And what else can I then do than get dirty with your code, to test if it is right? If you suspicion about the CR72 is correct, then I really really wonder why not errata article is out there? This situation reminds me of my master thesis. I had to struggle long to find out the Forster distance calculation in Lakowicz was wrong. The bible of fluorescence... Jesus.... but thanks for the beer! Best Your biggest fan. ;-) 2014-05-04 22:59 GMT+02:00 Andrew Baldwin <andrew.baldwin@xxxxxxxxxxxxx>: Dear Troels, The existence of typos is exceedingly probable! The editors have added more errors in the transcrpition, so it'll be curious to see how accurate the final draft will be. Your comments come just in time for alterations. So I'll pay a pint of beer for every identified error. I didn't use that particular supplementary section to directly calculate anything, so this is the place in the manu where errors are most likely to occur. I do note the irony that this is probably one of few bits in the article that people are likely to read. 1) Yep - that's a typo. alpha- should definitely be KEG-KGE (score 1 pint). 2) Yep! zs should be zetas (score 1 pint). 3) This one is more interesting. Carver and Richard's didn't define a deltaR2. To my knowledge, I think Nikolai was the first to do that in his 2002 paper on reconstructing invisible states. So here, I think you'll find that my definition is the correct one. There is a typo in the Carver Richard's equation. I've just done a brief lit review, and as far as I can see, this mistake has propagated into every transcription of the equation since. I should note that in the manu in this section. Likely this has gone un-noticed as differences in R2 are difficult to measure by CPMG, instead R2A=R2B being assumed. Though thanks to Flemming, it is now possible to break this assumption: J Am Chem Soc. 2009 Sep 9;131(35):12745-54. doi: 10.1021/ja903897e. Also, in the Ishima and Torchia paper looking at the effects of relaxation differences: Journal of biomolecular NMR. 2006 34(4):209-19 I understand their derivations started from Nikolai's formula, which is exact, so the error wouldn't come to light there either, necessarily. To prove the point try this: numerically evaluate R2,effs over a big grid of parameters, and compare say Nikolai's formula to your implementation of Carver Richard's with a +deltaR2 and a -deltaR2 in the alpha- parameter. Which agrees better? You'll see that dR2=R2E-R2G being positive wins. The attached should also help. It's some test code I used for the paper that has been briefly sanitised. In it, the numerical result, Carver Richards, Meiboom, Nikolai's and my formula are directly compared for speed (roughly) and precision. The code added for Nikolai's formula at higher precision was contributed by Nikolai after the two of us discussed some very odd results that can come from his algorithm. Turns out evaluating his algorithm at double precision (roughly 9 decimal places, default in python and matlab) is insuffficient. About 23 decimal places is necessary for exactness. This is what is discussed in the supplementary info in the recent paper. In short, on my laptop: formula / relative speed / biggest single point error in s-1 over grid Mine 1 7.2E-10 Meiboom 0.3 9E7 Carver Richards 0.53 8 Nikolai (double precision 9dp) 3.1 204 Nikolai (long double 18dp) 420 3E-2 Nikolai (23 dp) 420 2E-7 Numerical 118 0 The error in Carver Richard's with the alpha- sign made consistent with the Carver Richard's paper gives an error of 4989 s-1 in the same test. Ie, it's a bad idea. Note that the precise calculations in python at arbitrary precision are slow not because of the algorithm, but because of the mpmath module, which deals with C code via strings. Accurate but slow. This code has a small amount of additional overhead that shouldn't strictly be included in the time. I've no doubt also that algorithms here could be sped up further by giving them a bit of love. Those prefaces aside, I'm surprised that your benchmarking last Friday showed Nikolai's to be so much slower than mine? In my hands (attached) at double precision, Nikolai's formula is only 3x slower than mine using numpy. Though this is not adequate precision for mistake free calculations. Perhaps more physically compelling, to get my equation to reduce to Carver Richard's, alpha- has to be defined as I show it. Note that the origin of this parameter is its role in the Eigenvalue of the 2x2 matrix. This is the one advantage of a derivation - you can see where the parameters come from and get a feel for what their meaning is. So this typo, and the fact that Carver and Richards inadvertently change their definition of tau_cpmg half way through the paper, are both errors in the original manuscript. Though given that proof reading and type-setting in 1972 would have been very very painful, that there are this few errors is still actually quite remarkable. I notice in your CR72 code, you are currently using the incorrect definition, so I would suggest you change that. So overall, I think you're still ahead, so I'm calling that 2 pints to Troels. I would actually be very grateful if you can bring up any other inconsistencies in the paper! Also from an editorial perspective, please feel free to note if there are parts of the paper that were particularly unclear when reading. If you let me know, there's still time to improve it. Thusfar, I think you are the third person to read this paper (the other two being the reviewers who were forced to read it), and you're precisely its target demographic! Best, Andy. On 03/05/2014 13:54, Troels Emtekær Linnet wrote: Dear Andrew Baldwin. I am in the process of implementing your code in relax. I am getting myself into a terrible mess by trying to compare equations in papers, code and conversions. But I hope you maybe have a little time to clear something up? I am looking at the current version of your paper: dx.doi.org/10.1016/j.jmr.2014.02.023 which still is still "early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form." 1) Equations are different ? In Appendix 1 – recipe for exact calculation of R2,eff h1 = 2 dw (dR2 + kEG - kGE) But when I compare to: Supplementary Section 4. Relation to Carver Richards equation. Equation 70. h1 = zeta = 2 dw alpha_ = 2 dw (dR2 + kGE - kEG) Is there a typo here? The GE and EG has been swapped. 2) Missing zeta in equation 70 There is missing \zeta instead of z in equation 70, which is probably a typesetting error. 3) Definition of " Delta R2" is opposite convention? Near equation 10, 11 you define: delta R2 = R2e - R2g if we let e=B and g=A, then delta R2 = R2B - R2A And in equation 70: alpha_ = delta R2 + kGE - kEG but i CR original work, A8, alpha_ = R2A - R2B + kA - kB = - delta R2 + kGE - kEG So, that doesn't match? Sorry if these questions are trivial. But I hope you can help clear them up for me. :-) Best Troels 2014-05-01 10:07 GMT+02:00 Andrew Baldwin <andrew.baldwin@xxxxxxxxxxxxx>: The Carver and Richards code in relax is fast enough, though Troels might have an interest in squeezing out a little more speed. Though it would require different value checking to avoid NaNs, divide by zeros, trig function overflows (which don't occur for math.sqrt), etc. Any changes would have to be heavily documented in the manual, wiki, etc. The prove that the two definitions are equivalent is relatively straightforward. The trig-to-exp command in the brilliant (and free) symbolic program sage might prove helpful in doing that. For the tau_c, tau_cp, tau_cpmg, etc. definitions, comparing the relax code to your script will ensure that the correct definition is used. That's how I've made sure that all other dispersion models are correctly handled - simple comparison to other software. I'm only aware of two definitions though: tau_cpmg = 1 / (4 nu_cpmg) tau_cpmg = 1 / (2 nu_cpmg) tau_cpmg = 1/(nu_cpmg). Carver and Richard's switch definitions half way through the paper somewhere. What is the third? Internally in relax, the 1 / (4 nu_cpmg) definition is used. But the user inputs nu_cpmg. This avoids the user making this mistake as nu_cpmg only has one definition. I've seen people use nu_cpmg defined as the pulse frequency. It's just an error that students make when things aren't clear. I've often seen brave student from a lab that has never tried CPMG before do this. Without someone to tell them that this is wrong, it's not obvious to them that they've made a mistake. I agree with you that this is solved with good documentation. You guys are free to use my code (I don't mind the gnu license) or of course implement from scratch as needed. Cheers! For a valid copyright licensing agreement, you'll need text something along the lines of: "I agree to licence my contributions to the code in the file http://gna.org/support/download.php?file_id=20615 attached to http://gna.org/support/?3154 under the GNU General Public Licence, version three or higher." Feel free to copy and paste. No problem: "I agree to licence my contributions to the code in the file http://gna.org/support/download.php?file_id=20615 attached to http://gna.org/support/?3154 under the GNU General Public Licence, version three or higher." I'd like to note again though that anyone using this formula to fit data, though exact in the case of 2site exchange/inphase magnetisation, evaluated at double floating point precision should not be doing so! Neglecting scalar coupling/off resonance/spin flips/the details of the specific pulse sequence used will lead to avoidable foobars. I do see value in this, as described in the paper, as being a feeder for initial conditions for a more fancy implemenation. But beyond that, 'tis a bad idea. Ideally this should appear in big red letters in your (very nice!) gui when used. In relax, we allow the user to do anything though, via the auto-analysis (hence the GUI), we direct the user along the best path. The default is to use the CR72 and 'NS CPMG 2-site expanded' models (Carver & Richards, and Martin Tollinger and Nikolai Skrynnikov's Maple expansion numeric model). We use the CR72 model result as the starting point for optimisation of the numeric models, allowing a huge speed up in the analysis. The user can also choose to not use the CR72 model results for the final model selection - for determining dispersion curve significance. Here's the supp from my CPMG formula paper (attached). Look at the last figure. Maybe relevant. Nikolai's algorithm blows up sometimes when you evaluate to double float point precision (as you will when you have a version in python or matlab). The advantage of Nicolai's formula, or mine is that they won't fail when Pb starts to creep above a per cent or so. Using the simple formula as a seed for the more complex on is a good idea. The most recent versions of CATIA have something analogous. As for the scalar coupling and spin flips, I am unaware of any dispersion software that handles this. CATIA. Also cpmg_fig I believe. In fact, I think we may have had this discussion before? https://plus.google.com/s/cpmg%20glove If you consider experimental validation a reasonable justification for inclusion of the effects then you might find this interesting: Spin flips are also quite relevant to NH/N (and in fact any spin system). The supplementary section of Flemming and Pramodh go into it here for NH/N http://www.pnas.org/content/105/33/11766.full.pdf And this: JACS (2010) 132: 10992-5 Figure 2: r2 0.97, rmsd 8.0 ppb (no spin flips) r2 0.99, rmsd 5.7 ppb (spin flips). The improvements are larger than experimental uncertainties. When we design these experiments and test them, we need to think about the details. This is in part because Lewis beats us if we don't. You can imagine that it comes as a surprise then when we see people neglecting this. In short, the parameters you extract from fitting data will suffer if the details are not there. In the case of spin flips, the bigger the protein, the bigger the effect. In your code, you have the opportunity to do things properly. This leaves the details behind the scenes, so the naive user doesn't have to think about them. And only Flemming's CATIA handles the CPMG off-resonance effects. This is all explained in the relax manual. I have asked the authors of most dispersion software about this too, just to be sure. I don't know how much of an effect these have though. But one day they may be implemented in relax as well, and then the user can perform the comparison themselves and see if all these claims hold. Myint, W. & Ishima, R. Chemical exchange effects during refocusing pulses in constant-time CPMG relaxation dispersion experiments. J Biomol Nmr 45, 207-216, (2009). also: Bain, A. D., Kumar Anand, C. & Nie, Z. Exact solution of the CPMG pulse sequence with phase variation down the echo train: application to R(2) measurements. J Magn Reson 209, 183- 194, (2011). Or just simulate the off-resonance effects yourself to see what happens. For NH you see the effects clearly for glycines and side chains, if the carrier is in the centre around 120ppm. The problem generally gets worse the higher field you go though this of course depends when you bought your probe. You seem to imply that these effects are almost mythical. I assure you that they come out happily from the Bloch equations. Out of curiosity, from a philosophical perspective, I wonder if you'd agree with this statement: "the expected deviations due to approximations in a model should be lower than the experimental errors on each datapoint." Anyway, the warnings about analytic versers numeric are described in the manual. But your new model which adds a new factor to the CR72 model, just as Dimitry Korzhnev's cpmg_fit software does for his multiple quantum extension of the equation (from his 2004 and 2005 papers), sounds like it removes the major differences between the analytic and numeric results anyway. In any case, I have never seen an analytic result which is more than 10% different in parameter values (kex specifically) compared to the numeric results. I am constantly on the lookout for a real or synthetic data set to add to relax to prove this wrong though. I think there's a misunderstanding in what I mean by numerical modelling. For the 2x2 matrix (basis: I+, ground and excited) from the Bloch McConnell equation, you can manipulate this to get an exact solution. Nikolai's approach does this, though his algorithm can trip up sometimes for relatively exotic parameters when you use doubles (see attached). My formula also does this. I agree with you entirely that in this instance, numerically solving the equations via many matrix exponentials is irrelevant as you'll get an identical result to using a formula. My central thesis here though is that to get an accurate picture of the experiment you need more physics. This means a bigger basis. To have off resonance effects, you need a minimum 6x6 (Ix,Iy,Iz, ground and excited). To include scalar coupling and off resonance, you need a 12x12 (Ix,Iy,Iz,IxHz,IyHz,IzHz, ground and excited). Including R1 means another element and so on. The methyl group, for example, means you need 24. So when we use the term numerical treatment, we generally mean a calculation in a larger basis, as is necessary to take into account the appropriate spin physics. There aren't formulas to deal with these situations. In the 6x6 case for example, you need 6 Eigenvalues, which is going to make life very rough for someone brave enough to attempt a close form solution. The Palmer and Trott trick used in 2002 for R1rho is a smart way of ducking the problem of having 6 Eigenvalues, but for CPMG unfortunately you need several Eigenvalues, not just the smallest. The 2x2 matrix that Nikolai and Martin, Carver Richard's and I analyse does not include scalar coupling, as magnetisation is held in-phase (in addition to neglecting all the other stuff detailed above). So it is a good representation for describing the continuous wave in-phase experiments introduced here (neglecting relaxation effects and off resonance): Vallurupalli, P.; Hansen, D. F.; Stollar, E. J.; Meirovitch, E.; Kay, L. E. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18473–18477. and here: Baldwin, A. J.; Hansen, D. F.; Vallurupalli, P.; Kay, L. E. J. Am. Chem. Soc. 2009, 131, 11939–48. But I think are the only two where this formula is directly applicable. Only if you have explicit decoupling during the CPMG period do you satisfy this condition. So this is not the case for all other experiments and certainly not true for those used most commonly. Anyhow. Best of luck with the software. I would recommend that you consider implementing these effects and have a look at some of the references. The physics are fairly complex, but the implementations are relatively straightforward and amount to taking many matrix exponentials. If you do this, I think you'd end up with a solution that really is world-leading. As it stands though, in your position, I would worry that on occasion, users will end up getting slightly wrong parameters out from your code by neglecting these effects. If a user trusts this code then, in turn, they might lead themselves to dodgy biological conclusions. For the time being, I'll stick to forcing my students to code things up themselves. All best wishes, Andy.
Attachment:
CR72_disp_1_N.png
Description: PNG image
Attachment:
CR72_kex_pA.png
Description: PNG image
Attachment:
CR72_dw_pA.png
Description: PNG image
Attachment:
CR72_dw_kex_pA.png
Description: PNG image
Attachment:
CR72_dw_kex.png
Description: PNG image
Attachment:
B14_pA_kex.png
Description: PNG image
Attachment:
B14_pA_dw.png
Description: PNG image
Attachment:
B14_kex_pA_dw.png
Description: PNG image
Attachment:
B14_dw_kex.png
Description: PNG image
Attachment:
B14_disp_1_N.png
Description: PNG image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}